http://dx.doi.org/10.7775/rac.v91.i1.20598
ORIGINAL ARTICLE
A Preliminary Study of the
Phenotype-Genotype Correlation in Cardiomyopathies in Patients Referred to a
Tertiary Healthcare Center in the Suburbs of Buenos Aires
Estudio preliminar de correlación fenotipo-genotipo en
miocardiopatías de pacientes derivados a un centro de alta complejidad del
conurbano bonaerense
Gisela M. StreitenbergerMTSAC, 1, Graciela R. Reyes1, Maria P.
Velazco1, Viviana Pasquevich1, Mariela De Santos1, Marcos Granillo Fernández1, Mauricio Potito1, Pablo Kociubinski2, Javier MarianiMTSAC, 1
1 Cardiology Service, Hospital de Alta
Complejidad en Red El Cruce Néstor Kirchner. Av
Calchaquí 5401, Florencio Varela, Provincia de Buenos Aires, Argentina.
2 Diagnostic and Imaging Treatment
Service - Cardiomagnetic Resonance Area. Hospital de Alta
Complejidad en Red El Cruce Néstor Kirchner. Av. Calchaquí 5401, Florencio
Varela, Provincia de Buenos Aires, Argentina.
Address for reprints:
Gisela Streitenberger - E-mail: gisestreitenberger@yahoo.com.ar
ABSTRACT
Background: Cardiomyopathies
are defined as a disorder of the myocardium in which the heart muscle is
structurally and functionally abnormal, in the absence of coronary artery
disease, hypertension (HT), valvular heart disease
and congenital heart disease. These diseases are relatively common and a major
cause of morbidity and mortality worldwide.
Although genetic testing is
recommended for family screening, lack of solid data on specific
genotype-phenotype associations has reduced its impact on clinical management.
Objectives: This study aims to
analyze the frequency of mutations in a population of patients with cardiomyopathy
referred to a tertiary healthcare center and to analyze the genotype-phenotype
correlation of the identified mutations.
Methods: We prospectively
included 102 patients with suspected familial hypertrophic cardiomyopathy
(HCM), 70 of which were index cases, from an ambispective
cohort of patients with cardiomyopathies treated in a tertiary healthcare
public hospital in the province of Buenos Aires, from January 2012 to August
30, 2022.
Results: Of 102 patients,
83 were considered affected. Of these, 31 were HCM and 52 were phenocopies, with no difference in prognosis. A genetic
study was carried out in 77 patients, of whom 57
presented recognizable mutations, in 80% of the cases coinciding with a Mayo
Score ≥3. Twenty-eight variants of uncertain significance were detected.
Conclusions: It was confirmed
that molecular testing guided by the Mayo Score provided high probability of
detecting mutations. Molecular testing proved to be important due to the
phenotypic and genotypic overlap in cardiomyopathies. Understanding the
causative genetic variant, nowadays, does not affect the clinical management of
most HCM patients, but is helpful in a small group of genes with treatment
options.
Key words: Cardiomyopathies -
Cardiomyopathy Hypertrophic/genetics - Sarcomeres - Genetic Association Studies
- Genetic Testing
RESUMEN
Introducción:
Las
miocardiopatías se definen como un trastorno del miocardio en el que el músculo
cardíaco es estructural y funcionalmente anormal, en ausencia de enfermedad
arterial coronaria, hipertensión arterial (HTA), enfermedad valvular y enfermedad
cardíaca congénita. Estas enfermedades son relativamente frecuentes, y suponen
una importante causa de morbimortalidad a nivel global.
Aunque el estudio genético se recomienda para el cribado
familiar, la falta de datos robustos sobre asociaciones genotipo-fenotipo
específicas ha reducido su impacto en el manejo clínico.
Objetivos:
El objetivo de
este estudio es analizar la frecuencia de mutaciones en una población de pacientes
con miocardiopatía derivados a un centro de alta complejidad y el análisis de
la correlación genotipo-fenotipo en las mutaciones identificadas.
Material
y métodos: Se
estudiaron en forma prospectiva 102 pacientes con sospecha de miocardiopatía
hipertrófica (MCH) familiar, de los cuales 70 constituían casos índices, de una
cohorte ambispectiva de pacientes con miocardiopatías
controladas en un hospital público de alta Complejidad de tercer nivel de
atención de la provincia de Buenos Aires, desde enero 2012 al 30 agosto 2022.
Resultados:
De 102 pacientes
83 fueron considerados afectados. De eelos, 31 eran
MCH y 52 fenocopias, sin diferencia en el pronóstico. Se realizó estudio
genético en 77 pacientes, de los cuales 57 presentaron mutaciones reconocibles,
en el 80% de los casos coincidentes con un Score de Mayo ≥3. Se
detectaron 28 variantes de significado incierto.
Conclusiones:
Se comprobó que
realizar estudio molecular guiado por el Score de Mayo permitió obtener un alto
grado de probabilidad de detectar mutaciones. Se evidenció la importancia del
estudio molecular debido a la existencia de solapamiento fenotípico y
genotípico de las miocardiopatías. El conocimiento de la variante genética
causal actualmente no afecta el manejo clínico de la mayoría de los pacientes
con MCH, pero es de ayuda ante un pequeño grupo de genes que tienen opciones de
tratamiento.
Palabras
clave: Cardiomiopatías
- Cardiomiopatía Hipertrófica - Sarcómeros - Estudio
de Asociación Genética - Pruebas Genética
Received: 02/12/2022
Accepted: 30/01/2023
INTRODUCTION
Cardiomyopathies are defined as a
disorder of the myocardium in which the heart muscle is structurally and
functionally abnormal, in the absence of coronary artery disease, hypertension
(HT), valvular heart disease and congenital heart
disease. These diseases are relatively common and a major cause of morbidity
and mortality worldwide. (1)
There are several classifications
which are intended to differentiate some cardiomyopathies from others, although
in many cases they are more confusing than helpful. (2,3)
Hypertrophic cardiomyopathy (HCM) is
a primary myocardial disease affecting both sexes, caused by mutations of genes
encoding sarcomere proteins, with an estimated prevalence of up to 1/200-500
people, often inherited, with a complex genetic and phenotypic expression and a
natural history. (4-7)
Thousands of mutations in more than
50 genes have been described to be associated with HCM; however, the frequency
of the mutations identified may vary in different studies, and data available
are scarce. (4-7)
The mechanisms by which sarcomere
variants result in the clinical phenotype have not yet been elucidated.
Sarcomere genes trigger changes in the myocardium which lead to hypertrophy and
fibrosis, a small and stiff ventricle with impaired systolic and diastolic
function despite the preserved left ventricular ejection fraction (LVEF).
Several features, such as abnormal intramural coronary arteries responsible for
ischemia and mitral valve abnormalities, appear to have no direct association
with the sarcomere variants. (4-7)
Patients lacking a pathogenic variant
are believed to have non-Mendelian HCM and probably
better prognosis than patients with sarcomere pathogenic mutations. Identifying
the genetic basis of HCM creates opportunities to understand how the disease develops
and how to disrupt its progression. (5)
Although genetic testing is
recommended for family screening, lack of solid data on specific
genotype-phenotype associations has reduced its impact on clinical management. (5)
This study aims to analyze the
frequency of mutations in a population of patients with cardiomyopathy
referred to a tertiary healthcare center and the genotype-phenotype correlation
of the identified mutations.
METHODS
Study population
We prospectively included 102
patients with suspected familial HCM, 70 of which were index cases, from an ambispective cohort of patients with cardiomyopathies
treated in a tertiary healthcare public hospital in the province of Buenos
Aires from January 2012 to August 30, 2022.
The diagnosis of HCM was obtained
according to the criteria of the WHO and the European Society of Cardiology
(ESC) Working Group on Myocardial and Pericardial Diseases. (1)
The patients diagnosed with HCM were
those presenting 1 major electrocardiographic (ECG) or transthoracic echocardiographic
(TTE) criterion or 2 minor TTE criteria plus 1 minor ECG criterion, or 2 minor
ECG criteria plus 1 minor TTE criterion. (7)
Patients diagnosed with
cardiomyopathy were asked about relevant medical and family history considering
three generations, underwent a physical examination, a genogram, an ECG, a TTE,
a Holter ECG (in affected patients), an exercise
stress test or a cardiopulmonary exercise test (in affected patients), a
complete blood count with NT-proBNP and troponin
counts (in affected patients).
Echocardiographic parameters
The studies were performed with an Epiq 7 CVx 3D echocardiograph
(Philips Medical Systems) using an S5-1 transducer. Measurements of LVEF and
diastolic function were obtained according to the recommendations of the
American Society of Echocardiography. (8,9)
Values of LVEF <52% in men and
<54% in women were considered depressed.
The Speckle-tracking analysis was
performed according to current EACI/ASE consensus recommendations. (10) Cine
loops from three standard LV apical views (four, two and three chambers) were
recorded using grey-scale harmonic imaging at the highest possible frame rate
(55-90 frames/s). The analysis of the recorded files was performed off-line by
an experienced echocardiographer unblinded
to the patient's diagnosis.
The 2D global longitudinal strain
(GLS) was evaluated in 16 LV segments on average (Strain post-processing software:
TOMTEC. Dynamic Heart Model). The operator manually
adjusted the region of interest in segments that could not be correctly traced.
Normal value of global GLS Philips: -21±2%.
Cardiac magnetic resonance (CMR)
A Philips Medical Systems Achieva X Series 3T machine was used. Anatomical images of
the heart were obtained with dark-blood and bright-blood sequences. A functional
study with triggered cine imaging was performed. Images were acquired by enhaced T1- and T2-weighted sequences, fat suppression,
sequences with variable TE and tagging. First-pass (0.1 mmol/kg)
and late images (late enhancement) sequences were obtained with gadolinium
injection (total dose: 0.2 mmol/kg), which were then
post-processed and assessed with Extended MR Space 2.6.3.3 software.
Mutational analysis
The Mayo HCM genotype predictor score
(Mayo Score) was used to predict the diagnostic yield of genetic testing and
guide the use of next generation sequencing (NGS) method. (11,12)
Molecular testing was performed on
those with Mayo Score ≥3 (range -1 to 5) or relatives of patients with
positive mutations. First-degree relatives underwent a physical examination,
an ECG and a TTE to identify those who were affected. They were offered to
undergo genetic testing.
The technical component of the
confirmatory sequencing was performed by Invitae
Corporation from saliva samples collected by oral swabbing. The classification
of identified variants was carried out pursuant to the guidelines of the
American College of Medical Genetics and Genomics. (13)
All patients signed an informed
consent form, and the study was approved by the institution's ethics committee.
Study of the genotype-phenotype
correlation
For the description of the phenotypic
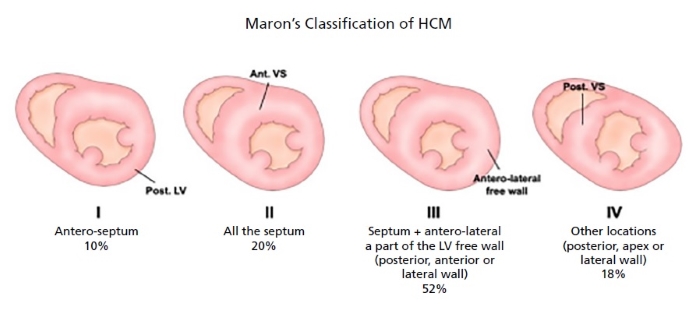
characteristics, the classical four phenotypes of Maron´s
classification based on the location and degree of hypertrophy was used. (14)
To correlate phenotype (F) with
genotype (G), the Lever’s classification was used to evaluate the pretest
probability based on the anatomical subtype. (15)
HCM was defined as obstructive when
it had a significant intraventricular pressure
gradient (≥30 mmHg) at rest, as latent obstructive when the gradient was
evident following provocation maneuvers (Valsalva/standing/exercise)
and as non-obstructive when the gradient was <30 mmHg.
The definitions are shown in the Appendix.
Cardiovascular events
Cardiovascular (CV) events were
defined as the presence or absence of the following:
• Need for implantable cardioverter defibrillator (ICD) or pacemaker implantation.
• Sudden death
• Cardiovascular hospitalization
• Septal myectomy or alcohol septal
ablation
The follow-up lasted 3 years
following the diagnosis by means of outpatient clinic visits or telephone
calls.
Statistical analysis
The statistical softwares
Epi Info for PC version 7.2.4.0 and Statistix 7 were used.
Qualitative variables were described
using numbers and percentages. Quantitative variables were described using mean
and standard deviation or median and interquartile range (IQR), depending on
normal or non-normal distribution, respectively.
For comparisons between groups,
Student's t test was used for continuous variables with normal distribution,
and non-parametric tests, such as Mann-Whitney U test for continuous variables
with non-normal distribution and Chi-square test (χ²) or Fisher's exact test for
categorical variables. A p-value <0.05 was considered statistically significant.
RESULTS
A total of 102 patients were
evaluated. The diagnostic flow diagram is shown in Figure 1.
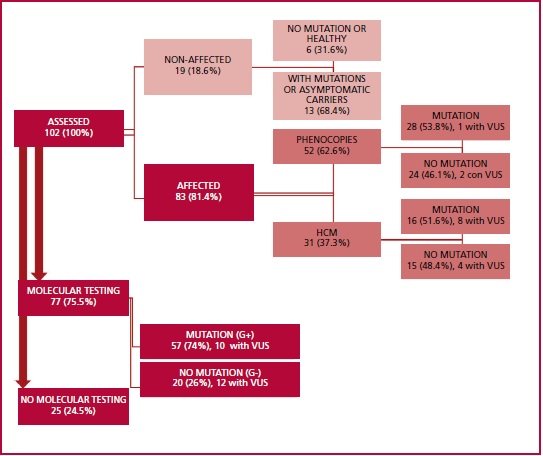
HCM: hypertrophic cardiomyopathy; VUS: variant of
uncertain significance
Fig. 1. Diagnostic flowchart
The presence of a phenotype
compatible with HCM was determined based on electrocardiographic and
echocardiographic criteria. Patients were classified into two groups:
"affected" (n= 83, 81.4%, 95% CI 72.4-88.4%)
and "non-affected" (n=19, 18.6%, 95% CI 11.6-27.5%) (Table
1).
Table 1. Studied population characteristics. A. Clinical and electrocardiographic parameters
|
Patients (N=102) |
Affected (n=83) |
Non-affected (n=19) |
p |
|
|
Current age (years) |
45 ± 16 |
47.6 ± 16 |
33.7 ± 10 |
<0.001 |
|
Age at diagnosis (years) |
41.9 ± 16 |
28.8 ± 14 |
0.001 |
|
|
Symptomatic |
83 (97.6%) |
81 (97.6%) |
2 (10.5%) |
<0.001 |
|
Female |
56 (54.9%) |
40 (48.2%) |
16 (84.2%) |
0.003 |
|
Family history |
71 (69.6%) |
52 (62.6%) |
19 (100%) |
<0.001 |
|
Weight (kg) |
70 ± 18 |
70 ± 18 |
69 ± 18 |
0.9 |
|
Hypertension |
24 (23.5%) |
23 (27.7%) |
1 (5.3%) |
0.02 |
|
Systolic blood pressure (mmHg) |
107 ± 16.8 |
105 ± 16 |
117 ± 14 |
0.005 |
|
Obesity |
22 (21.5%) |
18 (21.7%) |
4 (21%) |
0.6 |
|
Heart rate (beats/minute) |
71.6 ± 15 |
71.3 ± 16 |
73.2 ± 8.3 |
0.62 |
|
Diabetes |
8 (7.8%) |
7 (8.4%) |
1 (5.2%) |
0.53 |
|
Dyslipidemia |
10 (9.8%) |
10 (12%) |
0 |
0.11 |
|
Dyspnea NYHA Functional Classification
≥II |
80 (78.4%) |
80 (96.4%) |
0 |
<0.001 |
|
Angina |
38 (37.2%) |
37 (44.5%) |
1 (5.3%) |
<0.001 |
|
Syncope |
50 (49%) |
48 (57.8%) |
2 (10.5%) |
< 0.001 |
|
Coronary
artery disease |
4 (3.9%) |
4 (4.8%) |
0 |
0.43 |
|
BNP value (pg./mL) |
|
986 (122-3237) |
|
|
|
Troponin I |
|
12.5 (4-59.5) |
|
|
|
ELECTROCARDIOGRAM |
|
|
|
|
|
Abnormal |
88
(86.3%) |
82
(98.8%) |
6
(31.6%) |
|
|
Negative
T waves |
34 (33.3%) |
33 (39.7%) |
1 (5.3%) |
< 0.001 |
|
LVH
signs |
54
(52.9%) |
51
(61.4%) |
3
(15.8%) |
<
0.001 |
|
CLBBB
|
12 (11.7%) |
12 (14.4%) |
0 |
0.07 |
|
CRBBB |
4
(3.9%) |
4
(4.8%) |
0 |
0.43 |
|
LAHB |
23(22.5%) |
23 (27.7%) |
0 |
<0.001 |
|
QS
pattern |
52
(50.9%) |
51 (61.4%) |
1
(5.3%) |
<0.001 |
|
Microvoltage |
25 (24.5%) |
25 (30.1%) |
0 |
0.01 |
|
Sinus
rhythm |
92
(90.2%) |
73
(87.9%) |
19
(100%) |
0.24 |
|
Paroxysmal
or permanent AF |
18 (17.6%) |
18 (21.7%) |
0 |
0.01 |
|
Ventricular
tachycardia |
12
(11.7%) |
12
(14.4%) |
0 |
0.07 |
|
ECHOCARDIOGRAPHY |
|
|
|
|
|
Normal
LVEF |
78
(76.5%) |
59
(71%) |
19
(100%) |
<0.001 |
|
LVEF
(%) |
62 (55-66) |
60 (52-67) |
66 (61-70) |
0.03 |
|
LV
hypertrophy |
61
(59.8%) |
61
(73.5%) |
0 |
<0.001 |
|
LA
dilatation |
79 (77.4%) |
76 (91.5%) |
3 (15.8%) |
<0.001 |
|
Diastolic
dysfunction |
82
(80.4%) |
80
(96.4%) |
2
(10.5%) |
<0.001 |
|
E/e´
ratio |
11.6 ± 4.6 |
12.7 ± 4.2 |
6.6 ± 1.7 |
<0.001 |
|
Average
GLS (%) |
17
(12-20) |
16
(10-19) |
22
(20-22) |
<0.001 |
AF: atrial fibrillation; CLBBB: complete left bundle
branch block; CRBBB: complete right bundle branch block; GLS: global
longitudinal strain; LA: left atrium ; LAHB: left
anterior hemiblock; LVEF: left ventricular ejection
fraction; LVH: left ventricular hypertrophy; LVSF: left ventricular systolic
function; SBP: systolic blood pressure.
Qualitative variables are expressed as n (%) and
quantitative variables as mean ± standard deviation or median and interquartile
range.
The mean age of symptom diagnosis was
39±16.7 years, with earlier onset in women (34.7±15 years; p<0.001), those
with family history (36.7±16 years; p<0.001) and pathogenic TNNT2 variants.
Among those affected, the presence of
symptoms was more frequent in men (n=43) than women (n=40), and dyspnea was the
most frequent symptom.
ECG and TTE were performed in 100% of
patients and CMR in 64 (62.7%). CMR was not performed in some patients due to
claustrophobia, denial, or cardiac devices.
Table 1B shows the
characteristics of the 83 "affected" patients. A total of 71% (n=59)
had preserved LVEF compared to 100% in those non-affected; p<0.001. A total
of 37.4% (n=31) had HCM and 62.6% (n=52) were phenocopies.
In approximately half of the patients, the HCM was classified as obstructive
(51.6%), and most of them (25) had preserved LVEF (80%). The mean
GLS of patients with amyloidosis was 14% (9-17) and
statistically different (p=0.01) from that of patients with HCM, and the
LVEF/GLS index with a cut-off value ≥4.3±1.6 allowed to differentiate
amyloidosis from HCM (p<0.001), as in previous studies. (16)
Table 1. Studied population characteristics. B. Echocardiographic and cardiac magnetic resonance characteristics of
“affected” population
|
|
Affected n=83 |
HCM n=31 |
Phenocopy n=52 |
p |
|
|
Normal LVEF |
59 (71%) |
25 (80.6%) |
34 (65.4%) |
0.10 |
|
|
LVEF (%) |
60 (52-67) |
64 (55-69) |
58 (44-66) |
0.09 |
|
|
IVS thickness (mm) |
13 (9-17) |
18 (15-28) |
12 (9.5-15) |
<0.001 |
|
|
LVMI (g/m2) |
109
(78-141) |
138 (119-185) |
109 (86-133) |
0.001 |
|
|
LVOTO |
16 (19.3%) |
16 (51.6%) |
0 |
<0.001 |
|
|
LA dilatation |
76 (91.9%) |
30 (96.7%) |
46 (88.4%) |
0.18 |
|
|
LAVI (ml/m2) |
41 (32-56) |
51.5 (43-82.5) |
40 (36-55) |
0.003 |
|
|
80 (96.4%) |
31 (100%) |
49 (94.2%) |
0.24 |
||
|
1-
Prolonged relaxation |
8 (9.6%) |
0 |
8 (15.4%) |
0.1 |
|
|
2-
Pseudonormal |
57 (67.5%) |
24 (77.4%) |
32 (61.5%) |
0.1 |
|
|
3-
Restrictive |
11 (13.2%) |
3 (9.7%) |
8 (15.4%) |
0.1 |
|
|
4-
Monophasic |
8 (9.6%) |
4 (12.9%) |
3 (5.7%) |
0.1 |
|
|
E/e´ ratio |
12.7 ± 4.2 |
13.4 ± 3.9 |
12.3 ± 4.4 |
0.27 |
|
|
Subaortic
membrane |
2 (2.4%) |
2(6.4%) |
0 |
0.13 |
|
|
Bicuspid AV |
2 (2.4%) |
2(6.4%) |
0 |
0.13 |
|
|
Aortic regurgitation |
16 (19.3%) |
10 (32.2%) |
6 (11.5%) |
0.02 |
|
|
Mitral regurgitation |
63 (76%) |
26 (83.8%) |
37 (71.1%) |
0.19 |
|
|
Tricuspid regurgitation |
68 (82%) |
28 (90.3%) |
40 (77%) |
0.10 |
|
|
sPAP |
27 (0-39) |
33 (25-43) |
30 (0-40) |
0.15 |
|
|
Average GLS (%) |
16 (10-19) |
17 (13-19) |
15 (9-17) |
<0.001 |
|
|
LVEF/GLS |
3.7 (3.3-4.7) |
3.6 (3.2-4.2) |
3.9 (3.4-5.3) |
0.27 |
|
|
CARDIAC
MAGNETIC RESONANCE |
63 (75.9%) |
25 (80.6%) |
38 (73%) |
0.43 |
|
|
|
80 (62-96) |
85 (62-122) |
73 (62-90) |
0.24 |
|
|
LVEF (%) |
61 (44-71) |
70 (59-73) |
60 (40-67) |
<0.001 |
|
|
RVEF (%) |
72 (61-78) |
71 (64-81) |
72 (60-77) |
0.34 |
|
|
LGE |
44 (72.1%) |
18 (85.7%) |
26 (65%) |
0.07 |
|
GLS: global longitudinal strain; HCM: hypertrophic
cardiomyopathy; IVS: interventricular septum; LA:
left atrium; LAVI: left atrial volume index; LGE: late gadolinium enhancement;
LVEF: left ventricular ejection fraction; LVMI: left ventricular mass index;
LVOTO: left ventricular outflow tract obstruction; RVEF: right ventricular
ejection fraction;;; sPAP:
systolic pulmonary artery pressure
Qualitative variables are expressed as n (%) and
quantitative variables as mean ± standard deviation or median and interquartile
range.
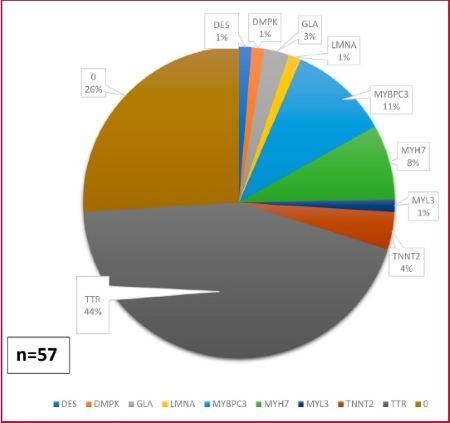
Identified mutations
A molecular testing was performed in
77 patients (75.5%), 57 of which (74%) had mutations (G+) and 20 (26%) did not
have (G-). Forty-six (80.7%) out of the 57 G+ patients had a Mayo Score ≥3 , p<0.001 vs. G- patients. Twenty-two (28.5%) variants
of uncertain significance (VUS) were detected (Figures 2 and 3). Two
patients had double heterozygosity pathogenic
variants and 10 had VUS in addition to the pathogenic mutation.
|
VARIANT |
n |
|
|
DES |
c.1360C>T - p.(Arg454Trp) |
1 |
|
DMPK |
605377: 19q13.32 |
1 |
|
GLA |
c.1244T>C p.(Leu415Pro) |
1 |
|
LMNA |
c.205del (p.Val69Trpfs*27) |
1 |
|
MYBPC3 |
c.1808_1821del (p.Ile603Thrfs*6) |
4 |
|
|
c.3192dup (p.Lys1065Glnfs*12) |
1 |
|
|
c.1624G>C (p.Glu542Gln) |
1 |
|
|
c.1877 C>G (p.Ser626) |
1 |
|
MYH7 |
c.485A>G (p.Tyr162Cys) |
1 |
|
|
c.788T>C (p.Ile263Thr) |
1 |
|
|
c.2770G>A (p.Glu924Lys) |
2 |
|
|
c.1208G>A (p.Arg403Gln) |
3 |
|
MYL3 |
c.454G>A - p.(Glu152Lys) |
1 |
|
TNNT2 |
c.812A>T (p.Asn271Ile) |
2 |
|
|
c.487_489del (p.Glu163del) |
1 |
|
TTR |
Thr60Ala |
1 |
|
|
Val50Met |
27 |
|
|
Val122Ile |
6 |
|
N |
TOTAL |
57 |
Fig. 2. Identified genetic variants
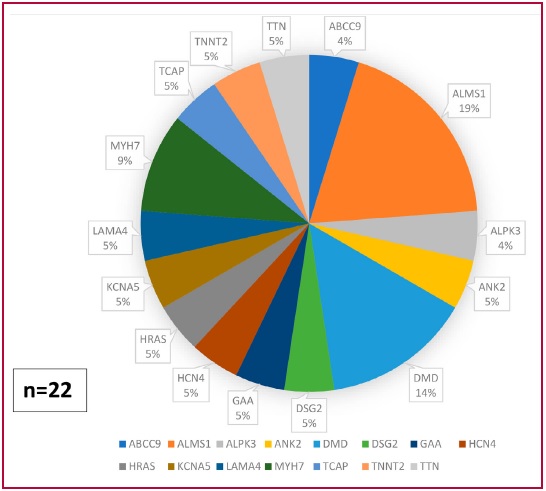
Fig. 3. Variants of uncertain significance (VUS)
Among the 19 non-affected patients,
disease could be ruled out in 6 (31.6%), while 13 (68.4%) were asymptomatic
carriers. Disease penetrance ("affected patients with mutations") was
77.2% (44 out of the 57 G+ patients); 16 (36.4%) out of the 44 had HCM.
Genotype-phenotype correlation
According to Maron’s
classification, the most frequent forms of presentation were type 1 and type 3
(septal and anterolateral involvement, respectively),
with more G+ detected in type 1 in HCM (9, 75%) and in type 3 in phenocopies (13, 68.4%), p<0.001.
Lever´s classification into 2 and 4
(reverse curvature septum and neutral septum, respectively) was useful when
assessing the likelihood of having G+ based on the anatomical phenotype
expressed by the patient (Table 2 and Figure 4).
Table 2. Genotype-phenotype correlation from patients who underwent molecular
testing
|
Patients n=77 |
|
With mutations (G+) n=57 |
|
Without mutations (G-) n=20 |
|
p |
|
|
Phenocopies |
42 (54.5%) |
|
33 (57.8%) |
|
9 (45%) |
|
0.46 |
|
HCM |
21 (27.3%) |
|
16 (28%) |
|
5 (25%) |
|
0.52 |
|
Maron 1 |
17 (22%) |
|
13 (23%) |
|
4 (20%) |
|
0.53 |
|
Maron 2 |
6 (7.8%) |
|
5 (8.7%) |
|
1 (5%) |
|
0.88 |
|
Maron 3 |
21 (27.3%) |
|
14 (24.6%) |
|
7 (35%) |
|
0.26 |
|
Maron 4 |
6 (7.8%) |
|
5 (8.7%) |
|
1 (5%) |
|
0.88 |
|
Lever 1 |
1 (1.3%) |
|
0 |
|
1 (5%) |
|
0.39 |
|
Lever 2 |
18 (23.4%) |
|
14 (24.5%) |
|
4 (20%) |
|
0.46 |
|
Lever 3 |
1 (1.3%) |
|
1 (1.75%) |
|
0 |
|
0.39 |
|
Lever 4 |
25 (32.4%) |
|
17 (29.8%) |
|
8 (40%) |
|
0.39 |
|
Age at diagnosis |
38 (29-48) |
|
36 (28-44) |
|
45.5 (37.5-51.5) |
|
0.05 |
|
Female |
46 (59.7%) |
|
36 (63.1%) |
|
10 (50%) |
|
0.44 |
|
HT |
15 (19.5%) |
|
8 (14%) |
|
7 (35%) |
|
0.04 |
|
CV events |
33
(42.8%) |
|
22
(38.6%) |
|
11 (55%) |
|
0.31 |
|
Heart transplant |
1 (4%) |
|
1 (2.2%) |
|
0 |
|
0.44 |
|
Ventricular tachycardia |
14
(13.8%) |
|
5
(8.7%) |
|
3 (15%) |
|
0.34 |
|
ICD |
16 (20.8%) |
|
11 (19.3%) |
|
5 (25%) |
|
0.40 |
|
Pacemaker |
12
(15.6%) |
|
8
(14%) |
|
4 (20%) |
|
0.37 |
|
Myectomy |
6 (7.8%) |
|
4 (7%) |
|
2 (10%) |
|
0.49 |
|
CV hospitalization |
26
(33.7%) |
|
16
(28%) |
|
10 (50%) |
|
0.06 |
|
CV death |
11 (14.3%) |
|
8 (14%) |
|
3 (15%) |
|
0.58 |
CV: cardiovascular; HT: hypertension; ICD: implantable
cardioverter defibrillator;
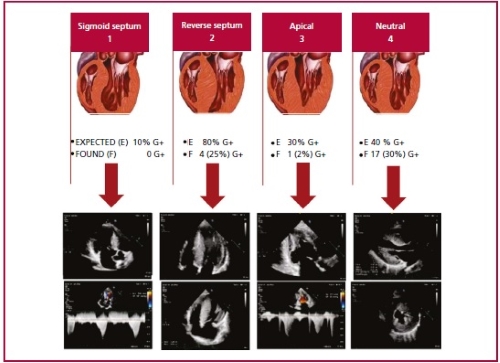
Color Doppler echocardiography image from own source.
Figure modified from Lever HM, Karam RF, Currie PJ,
Healy BP. Hypertrophic cardiomyopathy in the elderly. Distinctions from the
Young based on cardiac shape. Circulation 1989; 79(3):580-9.
Fig. 4. Hypertrophy patterns according to the classification of Lever et al.
Pretest probability for a positive genetic testing result according to the
anatomical subtype.
There were no significant differences
in CV events, medical treatments or procedures performed between G+ and G-
patients (Figure 5).
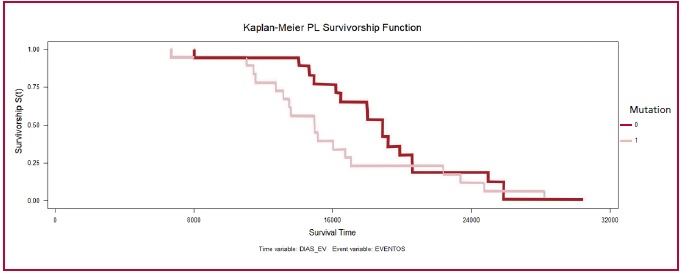
Fig.
5. Kaplan-Meier event-free survival curve. Patients with G+
have a mortality hazard ratio (HR) of 1.67 compared to those with G-, p=0.14.
The median time from the
cardiomyopathy onset to the CV event was 2.4 years. Patients with phenocopies (29, 55.7%) experienced the same number of CV
events as those with HCM (17, 54.8%), p=0.93. More cardiovascular deaths
occurred in men (10, 21.7%) than women (3, 5.3%); p=0.01.
DISCUSSION
Cardiomyopathies are a heterogeneous
group of myocardial diseases associated with mechanical and/or electrical dysfunction
that usually present with inappropriate ventricular hypertrophy or dilatation
and are due to a variety of causes, frequently genetic. (1)
For genetics to become useful in
clinical decision-making, it is necessary to get detailed information about the
clinical and morphological characteristics of the carriers of different
mutations, such as that provided by this study.
The Mayo Score enabled a better
selection of probands and a cost-effective molecular
testing, with a clear financial limitation.
Hypertrophic remodeling also occurs
in disorders that clinically mimic HCM, including Fabry’s
disease (mutations in GLA) and transthyretin (TTR)
amyloidosis, among others. More than 1500 mutations in at least 8 sarcomere
protein genes have been reported in HCM, although most (80%) mutations alter
the β-myosin heavy
chain (MYH7) or the myosin-binding protein C gene (MYBPC3). The diverse
molecular origin combined with the background genomic variability and lifestyle
differences between patients have made it difficult to definitively understand
the genotypic and phenotypic relationships. (4)
Previous studies suggest that
mutations in MYH7 cause about 15-30% of HCM cases. (17,18) In our patients, mutations in this
gene are less frequent and appear in 7.5% of the studied families. This
difference may be related to the degree of selection of the studied population.
Although we found no significant
differences in age at diagnosis, the mean age in patients with G+ was 37.4±15
years compared to 42.4±18 years in patients with G-, similar to other series. (19,20)
An interesting finding in our study
was the higher frequency of mutations identified in women (36, 63%; p=0.06),
without statistical significance; but as HCM is usually inherited in an autosomal
dominant manner, it would be expected that 50% of patients were female.
However, in nearly all series described, the proportion of women is about
30-40%, and they are usually older at the time of diagnosis. In our study, it
was the opposite (age at diagnosis in women: 37±15 years compared to 46.5±15
years in men; p<0.01). (17-20)
Women had a higher prevalence of the
obstructive phenotype, more severe symptoms requiring septal
reduction therapy and one patient even underwent a transplant. However, there
was higher mortality in men than women.
The identification of mutations in
different families allows a more accurate assessment of the genotype-phenotype
correlation and the proper interpretation of the pathogenic role of each mutation.
Several findings from our study emphasize the importance of a complete family
study. While in some mutations, such as TNNT2 c.812A>T (p.Asn271Ile), the
phenotype is reproduced similarly in most carriers, in others, such as MYBPC3
c.1808_1821del (p.Ile603Thrfs*6), there is a remarkable difference between the
phenotype of index cases (severe hypertrophy in young patients) and family
carriers with mild hypertrophy, in spite of having similar or older ages. In
these cases, it should be considered that additional genetic or environmental
factors may account for the large difference in expression. Several studies
have shown that HCM patients may have more than one mutation and that the
presence of double mutations is associated with a more severe expression of the
disease, as was the case in two patients in our study. (6, 17-20)
In clinical practice, HCM frequently
coexists with hypertension (8 patients with HCM in our study, 25.8%) due to
the high prevalence of both diseases. This hemodynamic situation inevitably
modifies the HCM phenotype as well as exercise (athlete's heart) and other
comorbidities (diabetes mellitus, obesity, and chronic renal failure). It is
clear that it is often impossible to recognize the real cause or the main
modifier of LV hypertrophy.
Nowadays, it is believed that
wild-type (wt) TTR amyloidosis (wtTTR),
which has been intensively studied and underdiagnosed, has relatively high
prevalence. In our study we detected 46 patients with cardiac amyloidosis (45%),
60.7% of which were TTR amyloidosis variant (TTRv),
26% wtTTR, and the rest other types of amyloidosis.
We believe that amyloidosis was a contributor to higher mortality in the "phenocopies" group in comparison with the HCM group.
Limitations
It was a single-center study
conducted at a tertiary healthcare center in individuals who were likely to be
more severely ill and symptomatic.
There may be some additional
mutations that have not been identified because of the use of predetermined
gene panels.
Samples are impossible to be
collected in deceased subjects or in those who declined to participate in the
study or were not notified to be an index case.
CONCLUSIONS
It was confirmed that molecular
testing guided by the Mayo Score provided high probability of detecting mutations.
Molecular testing proved to be important due to the phenotypic and genotypic
overlap in cardiomyopathies.
Understanding the causative genetic
variant does not currently affect the clinical management of most HCM patients,
but it is helpful in a small group of genes, such as GAA, GLA, LAMP2, PRKAG2
and TTR, which are undoubtedly associated with diseases that mimic HCM and have
different clinical profiles, inheritance patterns and treatment options;
therefore, in those cases, molecular testing represents a significant step
towards customized approaches.
Conflicts of interest
None declared.
(See authors conflicts of interest forms in the
website/ Supplementary material)
1. Elliott P, Andersson
B, Arbusteini, Bilinska Z, Cecchi F, Charron P, et al.
Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial
and pericardial diseases, European Heart Journal, Volume 29, Issue 2, January
2008, Pages 270–276, https://doi.org/10.1093/eurheartj/ehm342
2. Thiene
G, Corrado D, Basso C, Revisiting definition and
classification of cardiomyopathies in the era of molecular medicine, European
Heart Journal, Volume 29, Issue 2, January 2008, Pages 144–146, https://doi.org/10.1093/eurheartj/ehm585
3. Maron B, Desai
M, Nishimura R, et al. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy. J Am Coll Cardiol. 2022 Feb, 79 (4)
372–389. https://doi.org/10.1016/j.jacc.2021.12.002
4. Ommen
SR, Mital S, Burke MA, Day SM, Deswal
A, Elliott P, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of
patients with hypertrophic cardiomyopathy: a report of the American College of
Cardiology/American Heart Association Joint Committee on Clinical Practice
Guidelines. Circulation. 2020;142:e558–e631.
doi:
10.1161/CIR.0000000000000937
5. Mazzarotto
F, Olivotto I, Boschi B, Girolami F, Poggesi C, Barton
PJR, et al. Contemporary Insights Into the Genetics of Hypertrophic
Cardiomyopathy: Toward a New Era in Clinical Testing? J Am Heart Assoc. 2020;
0:e015473. DOI: 10.1161/JAHA.119.015473
6. Maron
BJ, Maron MS, Semsarian C,
Genetics of Hypertrophic Cardiomyopathy After 20
Years: Clinical Perspectives, Journal of the American College of Cardiology.
2012: 60 (8): 705-715, ISSN 0735- 1097, https://doi.org/10.1016/j.jacc.2012.02.068
7. McKenna WJ, Spirito
P, Desnos M, Dubourg O, Komajda M. Experience from clinical genetics in hypertrophic
cardiomyopathy: proposal for new diagnostic criteria in adult members of affected
families. Heart 1997;77(2):130-132
8. Lang RM, Badano
LP, Mor-Avi V, Afilalo J,
Armstrong A, Ernande L et al. Recommendations for
cardiac chamber quantification by echocardiography in adults: an update from
the American Society of Echocardiography and the European Association of
Cardiovascular Imaging. J Am Soc Echocardiogr.
2015; 28:1-39.
9. Nagueh
SF, Smiseth O A, Appleton CP, et al. Recommendations
for the evaluation of left ventricular diastolic function by echocardiography:
an update from the American Society of Echocardiography and the European
Association of Cardiovascular Imaging. J Am Soc Echocardiogr 2016; 29:277-314.
10. Voigt JU, Pedrizzetti
G, Lysyansky P, et al. Definitions for a common
standard for 2D speckle tracking echocardiography: consensus document of the
EACVI/ASE/Industry Task Force to Standardize Deformation Imaging. J Am Soc Echocardiogr 2015; 28:
183-93.
11. Bonaventura J, Norambuena P, Tomašov P, Jindrová D, Šedivá H, Macek M Jr, et.al. The utility of the Mayo Score for predicting the yield of genetic
testing in patients with hypertrophic cardiomyopathy. Arch Med Sci.
2019 May;15(3):641-649. doi: 10.5114/aoms.2018.78767. Epub
2018 Oct 8. PMID: 31110529; PMCID: PMC6524174.
12. Bos
J.M., Will M.L., Gersh B.J., Kruisselbrink
T.M., Ommen S.R., Ackerman M.J. Characterization of a
Phenotype-Based Genetic Test Prediction Score for Unrelated Patients With
Hypertrophic Cardiomyopathy. Mayo Clin.
Proc. 2014;89:727–737. doi: 10.1016/j.mayocp.2014.01.025.
13. Bennett RL, et al. Recommendations
for standardized human pedigree nomenclature. Pedigree
Standardization Task Force of the National Society of Genetic Counselors.
Am J Hum Genet. 1995 Mar;56(3):745-52.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1801187/
14. Maron
BJ. Hypertrophic cardiomyopathy. Curr
Probl Cardiol. 1993 Nov;18(11):639-704. doi:
10.1016/0146-2806(93)90025-w. PMID: 7903919.
15. Lever HM, Karam
RF, Currie PJ, Healy BP. Hypertrophic cardiomyopathy in the elderly.
Distinctions from the young based on cardiac shape. Circulation 1989 Mar;
79(3):580-9.
16. Saad A, Arbucci
R, Rousse G, Darú V, Merlo
P, Lowenstein J y col. Perfiles ecocardiográficos
del strain 2D permiten diferenciar a la amiloidosis cardíaca de la miocardiopatía hipertrófica con
fracción de eyección conservada. Revista argentina de cardiología, 2018:
86(6), 20-26. Recuperado en 23 de octubre de 2022, de http://www.scielo.org.ar/scielo.php?script=sci_arttext&pid=S1850-37482018000600020&lng=es&tlng=es
17. Maron
BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn
HJ, Seidman CE, et al. ACC/ESC clinical expert
consensus document on hypertrophic cardiomyopathy: a report of the American
College of Cardiology Task Force on Clinical Expert Consensus Documents and the
European Society of Cardiology Committee for Practice Guidelines (Committee to
Develop an Expert Consensus Document on Hypertrophic Cardiomyopathy). Eur Heart J, 24
(2003), pp. 1965-9
18. Laredo R, Monserrat L, Hermida-Prieto
M, Fernández X, Rodríguez I, Cazón L, et al. Mutaciones en el gen de la cadena
pesada de la betamiosina en la miocardiopatía
hipertrófica. Rev Esp
Cardiol. 2006;59(10):1008-18
19. Richard P, Charron
P, Carrier L, Ledeuil C, Cheav
T, Pichreau C, et al. Hypertrophic cardiomyopathy:
distribution of disease genes, spectrum of mutations, and implications for a
molecular diagnosis strategy. Circulation, 107 (2003), pp. 2227-32 http://dx.doi.org/10.1161/01.CIR.0000066323.15244.5
20. García-Castro M, Cotoa E, Reguerob JR, Berrazuetac JR, Álvareza V, Alonso B, et al. Mutaciones en genes sarcoméricos en la miocardiopatía hipertrófica. Rev Esp Cardiol.
2009;62(1):48-5.
Diagnostic criteria for HCM. World Health
Organization/International Society and Federation of Cardiology 1997
|
Major Criteria |
Minor Criteria |
|
Echocardiographic (TTE) |
|
|
- Anterior septum or
posterior wall ≥13 mm - Posterior septum or free
wall ≥ 15 mm - Severe SAM |
- Anterior septum or posterior wall ≥12 mm - Posterior septum or free wall ≥ 14 mm - Moderate SAM - Redundant mitral valve leaflets |
|
Electrocardiographic (ECG) |
|
|
- LVH + repolarization
changes - T wave inversion in
leads I and aVL, V3-V6 (≥3 mm), or II, III, aVF (≥5 mm) - Abnormal Q waves (>40
ms or >25% R wave) in at least 2 leads from II,
III, aVF, V1-4 or I aVL
or V5-6 |
- CLBBB or intraventricular conduction
defect in left ventricular leads - Minor repolarization changes in left ventricular leads - Deep S waves in V2 (>25 mm) |
|
Clinical |
Unexplained syncope, dyspnea, or precordial pain |
|
Diagnosis of hypertrophic cardiomyopathy |
|
|
1 major criterion, or |
2 minor TTE criteria
+ 1 minor ECG criterion |
SAM: systolic anterior motion; LVH: left ventricular
hypertrophy; CLBBB: complete left bundle branch block.
Table modified from McKenna WJ, Spirito
P, Desnos M, Dubourg O, Komajda M. Experience from
clinical genetics in hypertrophic cardiomyopathy: proposal for new diagnostic
criteria in adult members of affected families. Heart 1997;77(2):130-132Heart
1997;77(2):130-132
ESC Diagnostic Criteria for HCM 2008
Adults
Wall thickness ≥15 mm in one or
more LV segments – determined by any imaging technique: echocardiography,
cardiac magnetic resonance (CMR) or computed tomography (CT)–
that may not be explained by loading conditions only.
Children
LV wall thickening with a Z score >2 standard
deviations from the expected mean.
Family members
Unexplained presence of an increase in LV thickness
≥13 mm in one or more LV segments, determined by any imaging technique
(echocardiography, CMR or CT).
From Elliott P, Andersson
B, Arbustini E, Bilinska Z,
Cecchi F, Charron P, et al.
Classification of the cardiomyopathies: a position statement from the European
Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008;29(2):270-6
- HCM phenocopies
(imitations)": Cardiac or systemic diseases capable of producing LVH
that should not be labelled as HCM. The use of HCM to
describe increased LV wall thickness associated with systemic disorders or
secondary causes of LV hypertrophy (LVH) may be confusing. Systemic disorders
include metabolic and multi-organ syndromes, such as the RASopathies
(variants in several genes involved in RAS-MAPK signaling pathway),
mitochondrial myopathies, glycogen/lysosomal storage
diseases in children, and Fabry's cardiomyopathy,
amyloidosis, sarcoidosis, hemochromatosis and Danon's cardiomyopathy in adults. In these diseases,
although the magnitude and distribution of LV wall thickening may be similar to
those of the isolated HCM caused by variants in sarcomere genes, the
pathophysiological mechanisms responsible for the hypertrophy, the natural
history and the treatment strategies are different.
- Echocardiographic criteria for
amyloidosis were defined by the presence of LVH with a cut-off point ≥12
mm at the septal level or posterior wall according to
the 10th International Symposium on Amyloidosis, 2004.
|
Clinical variable |
Points |
|
Age <45 years |
1 |
|
Left ventricular wall
thickness >20 mm |
1 |
|
Family history of HCM |
1 |
|
Family history of sudden
cardiac death |
1 |
|
Reverse septal curvature |
1 |
|
Hypertension (HT) |
-1 |
Mayo HCM Genotype
Predictor Score
NGS results were compared to the Mayo
Score (range -1 to 5) according to clinical and echocardiographic variables.
One patient with a Mayo Score of 5 had a pathogenic mutation (100% yield). Patients with a Mayo Score of 4 had a pathogenic
mutation in 71% of the cases. Patients with a Mayo score of 3 or 2 had a
pathogenic mutation in 50% and 35% of the cases, respectively. The yield of
genetic testing with a score of -1 to 1 was low (6-21%).
Bonaventura J, Norambuena
P, Tomašov P, Jindrová D, Šedivá H, Macek M Jr, Veselka J. The
utility of the Mayo Score for predicting the yield of genetic testing in
patients with hypertrophic cardiomyopathy. Arch Med Sci. 2019 May;15(3):641-649. doi:
10.5114/aoms.2018.78767 Epub 2018 Oct 8. PMID: 31110529;
PMCID: PMC6524174.
ANNEX 2
Maron et al.9
have established a morphological classification into four types: type I, septal-anterior hypertrophy; type II, septal-anterior
and septal-posterior hypertrophy; type III, septal and anterolateral hypertrophy; and type IV, septal-posterior and/or anterolateral hypertrophy.
Modified from Maron BJ. Hypertrophic cardiomyopathy. Curr Probl Cardiol. 1993 Nov;18(11):639-704. doi:
10.1016/0146-2806(93)90025-w. PMID: 7903919.
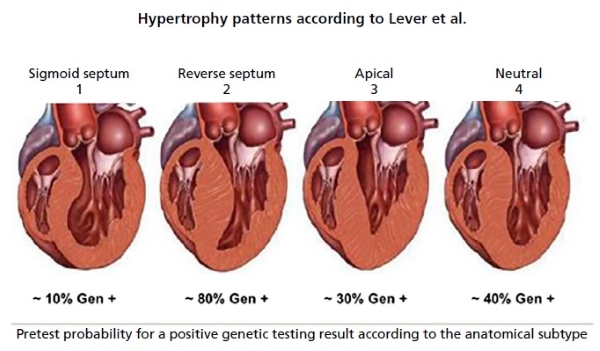
This classification has proved to be
very useful when considering the probability of presenting a positive genetic
testing based on the anatomical phenotype expressed by the patient, the
so-called echocardiography-guided genetic testing.
Modified from Lever HM, Karam
RF, Currie PJ, Healy BP. Hypertrophic cardiomyopathy in the elderly. Distinctions
from the young based on cardiac shape. Circulation 1989 Mar; 79(3):580-9.
ANNEX 3
Examples of clinical cases:
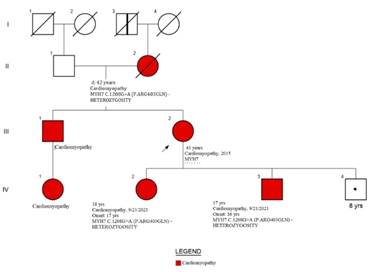
Example of a studied family’s genogram. References:
squares: men, circles: women, red: patients with clinical diagnosis of HCM,
white: patients without HCM or mutation or not assessed, symbols with central
black dot: mutation carriers without HCM phenotype, symbols with vertical black
bar: subjects with possible HCM by medical history (not proven). Diagonal line:
deceased patients, arrow: index case.
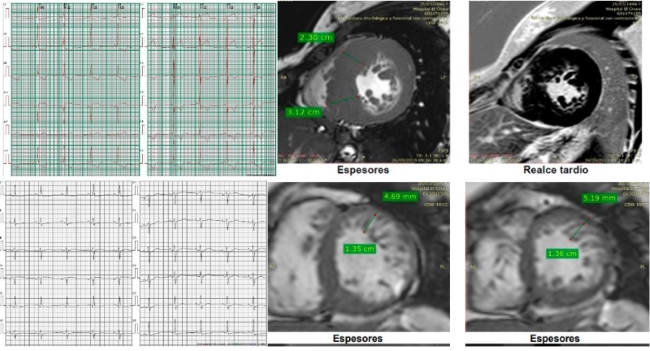
The index case is a woman with severe
hypertrophy diagnosed at the age of 37, with ICD implantation for syncopal sustained monomorphic ventricular tachycardia (VT)
at the age of 47. The CMR showed non-compacted myocardium (NCM) and G+ (MYH7:
c.1208G>A (p. Arg403Gln). Her 3 sons have G+, two of them with F+,
pathological ECG (left ventricular hypertrophy and negative T waves on the
anterolateral side). Above: ECG and CMR of one of her sons. Below: patient's
ECG and CMR.
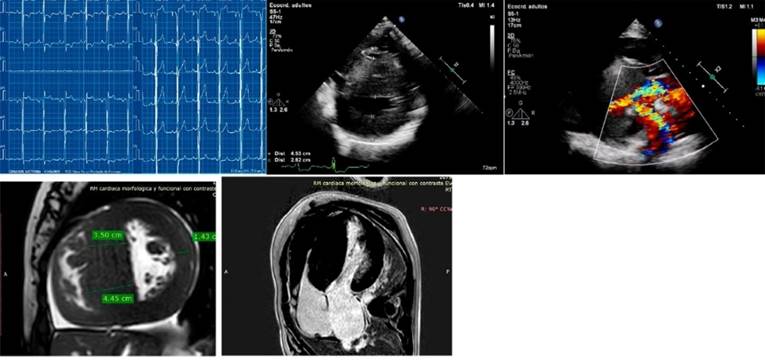
Case 2. A 17-year-old
adolescent with effort angina (F+ and G-) and VUS+.
ANNEX 4
Mutational analysis
On the basis of the saliva samples
collected by oral swabbing, the targeted regions of genomic DNA obtained from
the sample are enriched by hybridization-based protocol and sequenced by Illumina technology. All targeted regions are sequenced
with a depth ≥50x or supplemented with additional analyses.
Reads are aligned to a reference
sequence (GRCh37) and sequence changes are identified and interpreted in the
context of a single clinically relevant transcript, as listed below.
Enrichment and analysis focus on the
coding sequence of the indicated transcripts, 20 bp
of flanking intronic sequence and other specific
genomic regions shown to be responsible for the disease at the time of assay
design. Promoters, untranslated regions, and other
non-coding regions are not otherwise interrogated. For some genes, only
targeted loci (as indicated in the table above) are analyzed. Exonic deletions and duplications are called by an internal
algorithm that determines the number of copies at each target by comparing the
read depth for each target in the proband sequence
with the mean read depth and the read depth distribution, obtained from a
clinical dataset. Markers in the X and Y chromosomes are analyzed for quality
control purposes and may detect deviations from the expected sex chromosome
complement. Such deviations may be included in the report in accordance with
internal guidelines. Confirmation of the presence and location of reportable
variants is performed according to strict criteria established by Invitae (1400 16th Street, San Francisco, CA 94103, #
05D2040778), as necessary, using one of several validated orthogonal approaches
(PubMed ID 30610921). The following analyses are performed if relevant to the
request. For PMS2 exons 12-15, the reference genome was modified to force all
sequence reads derived from PMS2 and the PMS2CL pseudogene
to align to PMS2, and variant calling algorithms were modified to admit an
expectation of 4 alleles. If a rare SNP or indel
variant is identified by this method, both PMS2 and the PMS2CL pseudogene are amplified by long-range PCR and the location
of the variant is determined by Pacific Biosciences (PacBio)
SMRT sequencing of the relevant exon in both long-range amplicons.
If a CNV is identified, MLPA or MLPA-seq is run to
confirm the variant. If confirmed, both PMS2 and PMS2CL are amplified by
long-range PCR, and PacBio sequences the identity of
the fixed differences between PMS2 and PMS2CL from the long-range amplicon to disambiguate the location of the CNV.
The technical component of the
confirmatory sequencing is performed by Invitae
Corporation (1400 16th Street, San Francisco, CA 94103, #05D2040778). For the
C9orf72 repeat expansion testing, hexanucleotide
repeat units are detected by repeat primed PCR (RP-PCR) with fluorescently labelled primers, followed by capillary electrophoresis.
Interpretation reference ranges: benign (normal range): <25 repeat units,
uncertain: 25-30 repeated units, pathogenic (full mutation): ≥31 repeated
units. A second round of RP-PCR using a non-overlapping primer set is used to
confirm the initial call in the case of suspected allele sizes of 22 or more
repeats. For RNA analysis of the genes listed in the Genes Analyzed table,
complementary DNA is synthesized by reverse transcription from RNA derived from
a blood sample and enriched with specific gene sequences using capture
hybridization. After high-throughput sequencing with Illumina
technology, the output reads are aligned to a reference sequence (genome build
GRCh37; custom derivative of the RefSeq transcriptome) to identify the locations of exon junctions
through the detection of split reads. The relative usage of exon junctions in a
test sample is quantitatively assessed and compared to the usage observed in
control samples. Abnormal exonic junction usage is
evaluated as evidence in the Sherloc variant
interpretation framework. If an abnormal splicing pattern is predicted based on
a DNA variant outside the typical reportable range, as described above, the
presence of the variant is confirmed by targeted DNA sequencing. RNA sequencing
is performed by Invitae Corporation (1400 16th
Street, San Francisco, CA 94103, #05D2094793). Invitae
Corporation (5 Technology Drive, Irvine CA 92618, #05D1052995) performs the
technical component of fibroblast cell culture and gDNA
extraction from a skin punch biopsy.
A PMID is a unique identifier that
refers to a published scientific article. Search by PMID at http://www.ncbi.nlm.nih.gov/pubmed.
An rsID is
a unique identifier that refers to a single genomic position and is used to
associate population frequency information with sequence changes at that
position. The reported population frequencies are derived from several public
sites that aggregate data from large-scale population sequencing projects,
including ExAC (http://exac.broadinstitute.org), gnomAD (http://gnomad.broadinstitute.org) and dbSNP (http://ncbi.nlm.nih.gov/SNP).
A MedGen ID
is a unique identifier that refers to an article in MedGen,
NCBI's centralized database of information on genetic disorders and phenotypes.
Search by MedGen
ID at http://www.ncbi.nlm.nih.gov/medgen. An OMIM
number is a unique identifier that refers to a complete entry in the Online Mendelian Inheritance of Man (OMIM). Search by OMIM number
at http://omim.org/. Invitae
uses information from individuals undergoing testing to inform variant
interpretation. If "Invitae" is cited as a
reference in the variant details, this may refer to the individual in this
request and/ or to historical internal observations.