Rev Argent Cardiol 2024;92:231-234. http://dx.doi.org/10.7775/rac.v92.i3.20766
Dirección para correspondencia: Mariela Huertas. E-mail: marielahuertas92@gmail.com

Duchenne muscular dystrophy (DMD) is an X-linked genetic disorder that affects the expression of a protein essential for the stability of muscle fibers producing myocyte inflammation and death. Cardiac involvement is one of the main causes of morbidity and mortality in these patients. Most patients are asymptomatic or present with minimal symptoms because they self-limit their physical activity. (1,2) Cardiac involvement is characterized by progressive left ventricular dysfunction that progresses to dilated cardiomyopathy with subsequent heart failure, arrhythmias and sudden cardiac death.
We report the case of a 24-year-old male patient, with a history of muscle weakness, who visited the emergency department due to symptoms consistent with acute abdomen. The vital signs were normal. The physical examination showed a positive Murphy's sign. The ECG showed sinus rhythm, with poor R wave progression in precordial leads. The laboratory tests reported high white cell count and bilirubin levels. On abdominal ultrasound the gallbladder was distended and presented wall thickening. An 8.8 mm stone was observed in the infundibulum of the gallbladder and there was a 5.6 mm stone inside the gallbladder. The patient was admitted to the department of general surgery with a diagnosis of acute cholecystitis. He underwent laparoscopic cholecystectomy 48 hours later, with favorable outcome and was discharged 24 hours later.
Thereafter, after having visited the emergency department on two occasions due to lower extremity edema, he was admitted for diagnosis and monitoring with a clinical diagnosis of acute heart failure. A transthoracic echocardiography showed dilated cardiomyopathy with severe ventricular dysfunction, left ventricular ejection fraction (LVEF) of 29% with global hypokinesis. The patient was admitted to the coronary care unit (CCU) for further workup and treatment. During hospitalization, the patient reported a family history of Duchenne muscular dystrophy (DMD) and that he had undergone genetic diagnosis and muscle biopsy when he was 6 years old, which confirmed the diagnosis. However, the disease was irregularly followed-up and he had not been medicated.
The indications in the CCU included negative fluid balance, levosimendan pulses and complete medical treatment for advanced heart failure. During hospitalization an echocardiogram was performed with measurement of ventricular strain (-14%). There were no coronary artery lesions on coronary computed angiotomography with coronary calcium scoring. Cardiac magnetic resonance imaging (MRI) with gadolinium-based contrast agent demonstrated dilated cardiomyopathy with mid-myocardial delayed enhancement pattern involving the junction site between the right and left ventricular myocardium and anteroseptal and inferoseptal segments, and subepicardial enhancement of the anterolateral and inferolateral segments, a pattern also described in other entities such as idiopathic dilated cardiomyopathy and myocarditis. In addition, a 24-hour Holter monitoring was ordered, which only evidenced infrequent atrial premature contractions. After six days of hospitalization, the patient was discharged with indication for referral to a heart transplant center on an outpatient basis.
Figure 1
Color Doppler echocardiogram performed during hospitalization showing a LVEF of 29% (A) and mild tricuspid regurgitation (B); coronary computed angiotomography: the left anterior descending coronary artery has no lesions (C).
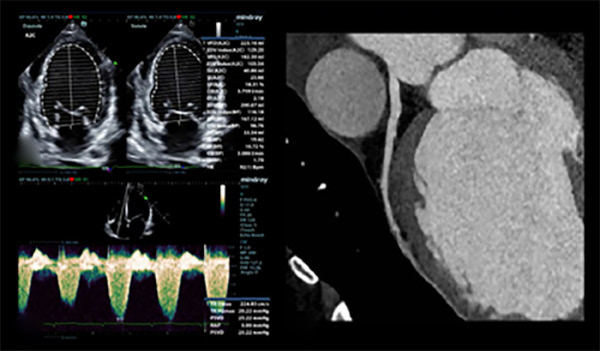
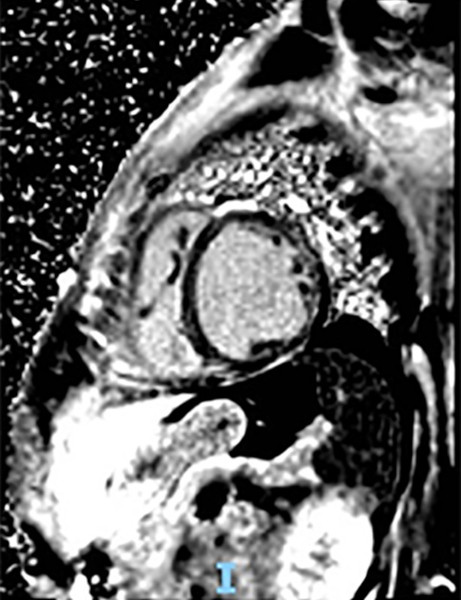
Figure 2
Cardiac magnetic resonance imaging with gadolinium-based contrast agent showing mid-myocardium delayed enhancement pattern involving the junction site between right and left ventricular myocardium and anteroseptal and inferoseptal segments, and subepicardial enhancement of the anterolateral and inferolateral segments.
One month later, the patient was hospitalized in a high-complexity center. As his clinical condition worsened, he was included on the emergency waiting list for transplantation. After being hospitalized for five months, the patient required ventricular assist device and remained on the emergency waiting list. Finally, heart transplantation was successfully performed, and the patient was discharged twenty-three days later. He is currently on post-transplant care and kinesiotherapy and receives genetic counseling with his family (mother and sister).
Becker muscular dystrophy and DMD are the most common muscular dystrophies in humans. They are X-linked genetic disorders with deletion, duplication and mutations of exons encoding for dystrophin, a protein that plays a role in the stabilization and signaling between myocyte membrane, extracellular matrix and cytoskeleton. While the genetic abnormality in Becker dystrophy consists of decreased expression of this protein or abnormal size, dystrophin is still functional. (1,3) In DMD, the genetic involvement leads to absence of expression, or expression of a non-functional protein, so it usually has a worse prognosis with a life expectancy of three decades. The latter variant of dystrophy is also the most common, with a prevalence of around 5 per 100 000 males and an incidence of 1 per 3800-6300 live-born males. (4)
Since the cardiac condition is due to an inherited myopathy, the therapeutic possibilities are limited. For the genetic analysis of our case we used the CTAB method, the DNA was analyzed by multiplex and simplex PCR to specifically find deletions, because of the loss of one or several exons of the dystrophin gene. Genomic deletion of exons 45 and 47 was detected, confirming the diagnosis of inconclusive muscular dystrophy: Duchenne/Becker.
Currently there are no clinical data or scientific evidence to support therapy, so each particular case should be evaluated by a Heart Team when considering heart transplantation, because although this is the standard of care for refractory heart failure, it is usually not recommended in patients with DMD because of comorbidities that limit the benefit. (1) However, there are case reports showing a favorable outcome in these patients.
DMD is a genetic, progressive, disabling and fatal disease that affects the skeletal muscle and requires early diagnosis, genetic counseling and the creation of a multidisciplinary team for treatment and comprehensive follow-up. The scope of this disease involves genetics and personalized medicine in the search of tailored treatments, although there is no definite treatment or cure yet. The clinical manifestations of heart failure occur in the end stages of the disease, therefore strict monitoring and follow-up is essential. (1,4,5,6)
Ethical considerations
Not applicable.
Conflicts of interest
Los autores declaran que no tienen conflicto de intereses. (Véanse formularios de conflicto de intereses de los au tores en la Web).
Financing
None
REFERENCES
3. Osorio AN, Cantillo JM, Salas AC, Garrido MM, Padilla JV. Consenso para el diagnóstico, tratamiento y seguimiento del paciente con distrofia muscular de Duchenne. Neurología 2019;34:469-81. https://doi.org/10.1016/j.nrl.2018.01.001
4. Finsterer J, Stöllberger C. Cardiac involvement in Becker muscular dystrophy. Can J Cardiol 2008;24:786-92. https://doi.org/10.1016/S0828-282X(08)70686-X