Rev Argent Cardiol 2024;92:236-238. http://dx.doi.org/10.7775/rac.es.v92.i3.20766
Dirección para correspondencia: Mariela Huertas. E-mail: marielahuertas92@gmail.com

La distrofia muscular de Duchenne (DMD) es un trastorno genético ligado al cromosoma X, que afecta la expresión de una proteína esencial para la estabilidad de las fibras musculares, por lo que su afección genera inflamación y muerte de las mismas. El compromiso cardíaco es una de las principales causas de morbimortalidad en estos pacientes. Gran parte de ellos cursan de forma asintomática u oligosintomática debido a la autolimitación al esfuerzo. (1,2) La afección cardíaca se caracteriza por una disfunción ventricular izquierda progresiva que evoluciona a miocardiopatía dilatada con el desarrollo de insuficiencia cardiaca, arritmias y muerte súbita.
Presentamos el caso de un joven de 24 años, con antecedente de debilidad muscular, que consulta por guardia externa por presentar cuadro compatible con abdomen agudo. Los signos vitales se encuentran dentro de la normalidad; al examen físico destaca signo de Murphy positivo. En el ECG presenta ritmo sinusal, con mala progresión de onda R en derivaciones precordiales. En el laboratorio leucocitosis e hiperbilirrubinemia. En la ecografía de abdomen se observa vesícula biliar distendida, de paredes engrosadas, con presencia de un lito de 8,8 mm en el bacinete y otro de 5,6 mm en su interior. Ingresa al servicio de Cirugía General con diagnóstico de colecistitis aguda, luego de 48 horas de internación se realiza colecistectomía laparoscópica, con buena evolución y alta sanatorial a las 24 horas.
Posteriormente consulta nuevamente por guardia externa en dos oportunidades por presentar edemas en miembros inferiores, por lo cual se decide su internación para diagnóstico y control. Se hace diagnóstico clínico de insuficiencia cardiaca aguda. Se solicita ecocardiografía transtorácica, que evidencia de miocardiopatía dilatada con deterioro grave de la función ventricular, fracción de eyección ventricular izquierda (FEVI) 29 %, con hipocinesia global debido a lo cual se decide su internación en Unidad Coronaria (UCO) para estudio y tratamiento. Durante la internación se logra recabar antecedente familiar y diagnóstico genético y biopsia muscular (realizados a los seis años de edad) de distrofia muscular de Duchenne (DMD), que ha tenido seguimiento irregular y sin tratamiento médico.
En UCO se indica balance hídrico negativo; pulso de levosimendan; y se inicia tratamiento médico completo para insuficiencia cardíaca avanzada. Durante la internación se realiza ecocardiograma con medición de strain (- 14 %); angiotomografía computada coronaria con score de calcio donde no se evidencian lesiones; resonancia magnética nuclear (RMN) cardíaca con gadolinio que constata miocardiopatía dilatada con patrón de realce tardío mesocárdico que involucra el sitio de unión entre miocardio ventricular derecho e izquierdo y anteroseptal e inferoseptal, además de realce subepicárdico anterolateral e inferolateral, patrón que también se encuentra descrito en otras entidades como la miocardiopatía dilatada idiopática y la miocarditis. Además se solicita ECG Holter de 24 hs que solo evidencia extrasistolia supraventricular de baja densidad. Luego de seis días de internación, se otorga el alta sanatorial con indicación de derivación a centro de trasplante de forma ambulatoria.
Figura 1
Ecocardiograma Doppler color realizado durante la internación donde se evidencia FEVI 29% (A) y jet de insuficiencia tricuspídea leve (B); angiotomografía coronaria, con arteria descendente anterior sin lesiones (C).
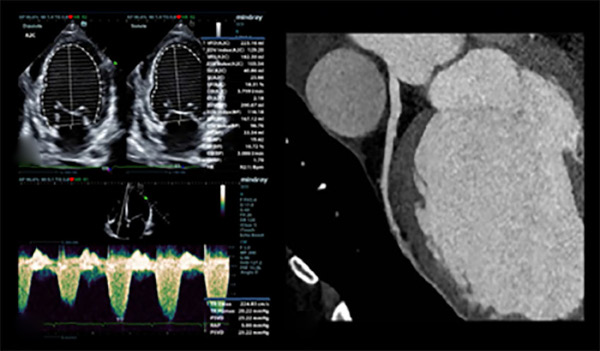
Figura 2
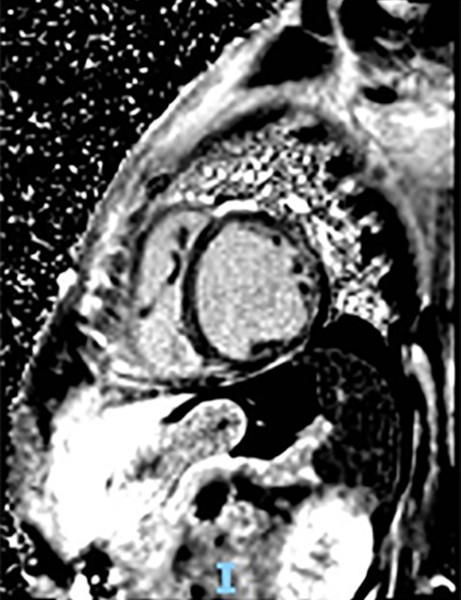
Resonancia magnética nuclear cardíaca con gadolinio. Se observa realce mesocárdico que involucra el sitio de unión entre miocardio ventricular derecho e izquierdo y anteroseptal e inferoseptal, y patrón de realce subepicárdico anterolateral e inferolateral.
Luego de un mes de evolución, se decide internación en un centro de mayor complejidad. Por deterioro del estado general, ingresa en lista de espera de urgencia para trasplante. Después de aproximadamente cinco meses de internación evoluciona con requerimiento de asistencia ventricular, y queda en lista de espera en emergencia. Finalmente se realiza trasplante cardíaco exitoso, con alta a los veintitrés días. Actualmente se encuentra cumpliendo cuidados postrasplante y rehabilitación kinésica motora. Recibe asesoramiento genético junto a su familia (madre y hermana).
La distrofia muscular de Becker y la DMD son las distrofias musculares más frecuentes en humanos. Consisten en enfermedades genéticas ligadas al cromosoma X, con deleción y/o duplicación, como así también mutaciones puntuales de exones que codifican para la expresión de la distrofina, una proteína que cumple funciones en la estabilización y señalización entre la membrana, matriz extracelular y el citoesqueleto del miocito. Mientras que la alteración genética en la distrofia de Becker consiste en la menor expresión de esta proteína o tamaño anormal, ésta sigue siendo funcional. (1,3) En la DMD, el compromiso genético lleva a la ausencia de expresión, o expresión de una proteína no funcional, por lo cual suele tener peor pronóstico con una expectativa de vida de tres décadas. Ésta última variante de distrofia es también la más frecuente, aproximadamente 5 por cada 100 000 varones y una incidencia de 1 por cada 3800-6300 recién nacidos varones. (4)
Debido a que la afección cardiaca se debe a una miopatía hereditaria, las posibilidades terapéuticas son limitadas. En cuanto al análisis genético del caso en cuestión, realizado por técnica CTAB, se analizó el ADN mediante PCR multiplex y simplex para hallar específicamente deleciones, producto de la pérdida de uno o varios exones del gen de la distrofina. Se detectó deleción genómica de los exones 45 y 47. Ello confirmó el diagnóstico de distrofia muscular no concluyente: Duchenne/Becker.
Actualmente no existen datos clínicos o evidencia científica que sustenten la terapéutica, por lo que cada caso en particular debe ser evaluado por un Heart Team al momento de plantear un trasplante, ya que si bien éste es el tratamiento estándar en insuficiencia cardíaca sin respuesta al tratamiento médico, se suele desestimar en pacientes con DMD por presentar comorbilidades que limitan el beneficio. (1) Sin embargo existen reportes de casos con evidencia de beneficio y buena evolución en estos pacientes.
La DMD es una patología genética, progresiva, discapacitante y mortal que compromete el músculo esquelético; requiere de un diagnóstico temprano, asesoramiento genético y la conformación de un equipo multidisciplinario para tratamiento como así también seguimiento en forma integral. Es una enfermedad que abarca desde la genética a la medicina individualizada en búsqueda de tratamientos individualizados, aunque no existe un tratamiento definido, ni curativo. La clínica de insuficiencia cardíaca se presenta en estadios terminales de la enfermedad, de modo que es imprescindible su control y seguimiento estrecho. (1,4,5,6)
Consideraciones Éticas
No aplica.
Declaración de conflicto de intereses
Los autores declaran que no tienen conflicto de intereses. (Véanse formularios de conflicto de intereses de los au tores en la Web).
Financiamiento
No se recibió financiamiento para la realización del presente trabajo
BIBLIOGRAFÍA
3. Osorio AN, Cantillo JM, Salas AC, Garrido MM, Padilla JV. Consenso para el diagnóstico, tratamiento y seguimiento del paciente con distrofia muscular de Duchenne. Neurología 2019;34:469-81. https://doi.org/10.1016/j.nrl.2018.01.001
4. Finsterer J, Stöllberger C. Cardiac involvement in Becker muscular dystrophy. Can J Cardiol 2008;24:786-92. https://doi.org/10.1016/S0828-282X(08)70686-X