Leonardo Caceres 1 MTSAC, Rodrigo Cano 1, Camila Correa Sadouet 1, Adrian Mahl 1, Gisela Streintenberger 1, Guillermo Mazo 1, Maribel Kanchi 1, Heraldo D’Imperio 1
1 Área de Investigación. Sociedad Argentina de Cardiología
Dirección para correspondencia: Leonardo Caceres. Área de Investigación de la Sociedad Argentina de Cardiología. E-mail: leo_1208_caceres@gmail.com
Rev Argent Cardiol 2024;92:147-153. http://dx.doi.org/10.7775/rac.es.v92.i2.20751
RESUMEN
Introducción: La miocardiopatía hipertrófica (MCH), es la enfermedad genética cardiovascular más común, causada por mutaciones en proteínas del sarcómero cardíaco, con una prevalencia considerable y clínica variable, desde asintomática hasta insuficiencia cardíaca y muerte súbita. Existen pacientes seguidos en centros no especializados, y es necesario conocer datos que puedan mostrar la realidad de su diagnóstico, tratamiento y pronóstico.
Objetivo: Conocer las características clínicas, estrategias diagnósticas y terapéuticas al abordar la MCH en centros no especializados en la patología.
Material y métodos: Estudio de corte transversal, multicéntrico, de alcance nacional, con análisis cuantitativo, de pacientes con MCH confirmada o altamente probable.
Resultados: Se registraron 95 pacientes, mayormente hombres con hipertensión arterial (40 %) y dislipidemia (22 %) como principales factores de riesgo. Se observó baja proporción de comorbilidades: enfermedad pulmonar obstructiva crónica (6 %), infarto de miocardio previo (5 %), accidente cerebro vascular previo (1 %) e insuficiencia renal crónica (1 %). Los síntomas principales fueron la disnea (47 %) y el ángor (27 %), y los métodos diagnósticos más usados fueron el ecocardiograma (97 %) y la resonancia cardíaca (71 %). La localización más frecuente fue septal, con 37 % de tipo obstructivo.
El test genético, realizado en un 33 %, fue positivo en más de la mitad de los pacientes. No se realizó en dos tercios de los casos principalmente por falta de cobertura.
Conclusiones: Los hallazgos son concordantes con los de registros internacionales. Con base a nuestros hallazgos, se resalta la necesidad de mejorar el acceso a estudios diagnósticos más complejos y optimizar recursos en un sistema de salud fragmentado.
Palabras clave: Miocardiopatía hipertrófica - Registro - Centros no especializados - Práctica clínica
ABSTRACT
Background: Hypertrophic cardiomyopathy (HCM) is the most common genetic disease caused by cardiac sarcomere protein mutations, with considerable prevalence and different clinical presentation, varying from asymptomatic to heart failure and sudden death. Some patients are followed-up in nonspecialized centers, and it is necessary to know data that show the reality of their diagnosis, treatment, and prognosis.
Objective: The aim of this study was to know the clinical characteristics, and diagnostic and therapeutic strategies when HCM is managed in centers not specialized in this disease.
Methods: This was a national, cross-sectional, multicenter study, with quantitative analysis of patients with confirmed or highly probable HCM.
Results: A total of 95 patients were recruited, mostly men, with hypertension (40%) and dyslipidemia (22%) as main risk factors. A low proportion of comorbidities was observed: chronic obstructive pulmonary disease (6%), prior myocardial infarction (5%), prior stroke (1%) and chronic kidney failure (1%). The main symptoms were dyspnea (47% and angina (27%), and the most used diagnostic methods were echocardiogram (97%) and cardiac magnetic resonance imaging (71%)). The most frequent localization was septal, with 37% of hypertrophic obstructive cardiomyopathy.
The genetic test, performed in 33% of patients, was positive in more than half of cases. It was not performed in the rest of the patients, mainly due to lack of health coverage.
Conclusions: These findings are in agreement with international registries. Based on our findings, emphasis should be placed in improving the access to more complex diagnostic studies and optimizing the resources in a fragmented health system.
Key words: Hypertrophic cardiomyopathy - Registry - Nonspecialized centers - Clinical practice
Recibido: 20/12/2023
Aceptado: 05/02/2024
INTRODUCCIÓN
La miocardiopatía hipertrófica (MCH) es la enfermedad genética con afectación cardiovascular más frecuente, con una prevalencia aproximada de 1 en 200/500. (1,2) Su etiología radica en la mutación de una serie de genes que codifican proteínas para el sarcómero cardíaco, con el desarrollo de hipertrofia del ventrículo izquierdo (HVI), desorden miofibrilar y fibrosis miocárdica. (3,4). Las manifestaciones clínicas son variables, y van desde asintomaticas a insuficiencia cardiaca y la muerte súbita. (5,6)
Esta entidad es un verdadero desafío diagnóstico debido a que existen miocardiopatías infiltrativas que se comportan como fenocopias. (5,7)
Actualmente se recomiendan tratamientos farmacológicos, implante de dispositivos y cirugías que han mejorado la sobrevida y calidad de vida. (8-10)
Existe un número importante de pacientes con sospecha de MCH que son seguidos en centros no especializados en la patología. Consideramos que los datos derivados de estos pacientes pueden contribuir a reflejar en forma más abarcativa la realidad de la enfermedad.
Presentamos un primer reporte del Registro de Miocardiopatía Hipertrófica en centros no especializados, actualmente en curso, en Argentina.
OBJETIVOS
Conocer las características clínicas de los pacientes y las estrategias diagnósticas y terapéuticas que aplican los cardiólogos clínicos en centros no especializados ante los cuadros altamente compatibles con MCH.
MATERIAL Y MÉTODOS
Se trata de un estudio observacional, retrospectivo y multicéntrico de alcance nacional, en pacientes con MCH confirmada o altamente probable. Participan cardiólogos clínicos que prestan atención en consultorios ambulatorios de centros no especializados.
Se incluyeron pacientes mayores de 18 años con diagnóstico confirmado o altamente compatible con MCH establecido por estudios de imágenes o laboratorio (ecocardiografía Doppler, resonancia cardíaca con gadolinio y/o test genético), a juicio de los cardiólogos clínicos que los tenían en seguimiento. Se excluyeron pacientes con patologías o situaciones que generan secundariamente hipertrofia ventricular: hipertensión arterial (HTA), valvulopatías, miocardiopatías infiltrativas, deportistas, etc.; y pacientes con MCH en seguimiento en centros especializados. Se definió como centro especializado a centros monovalentes de cardiología, y/o consultorios específicos de miocardiopatías.
La recolección de datos se realizó a través de la plataforma REDCap de la Sociedad Argentina de Cardiología y se realizó un corte desde el 1 de junio al 30 de septiembre de 2023.
Análisis estadístico
Las variables cualitativas se presentan como frecuencias y porcentajes. Para la descripción de las variables cuantitativas, se utilizó la media ± la desviación estándar (DE) o la mediana y el rango intercuartílico (RIC 25-75), según su distribución. El análisis de las variables discretas se realizó mediante el test de chi cuadrado o el de Fisher, según correspondiera, y el de las variables continuas con el test de t o el de Mann Whitney. Se consideró significativo un valor de p <0,05 a dos colas. El análisis se realizó con el software estadístico R.
Consideraciones éticas
El protocolo fue aprobado por el Comité en investigación del Gobierno de la Ciudad de Buenos Aires. No se tomó consentimiento informado por tratarse de un estudio con datos anonimizados sin datos personales.
RESULTADOS
Se incluyeron 95 pacientes distribuidos en 8 provincias. La edad media fue de 50 años con predominio del sexo masculino (58 %). La prevalencia de factores de riesgo cardiovascular fue: HTA 40 %, dislipidemia 22 %, obesidad 15 % y diabetes 14 %. Se observó baja proporción de comorbilidades: enfermedad pulmonar obstructiva crónica 6 %, infarto de miocardio previo 5 %, accidente cerebro vascular previo 1 % e insuficiencia renal crónica 1 % (Tabla 1).
Un tercio de los pacientes consultó por primera vez con diagnóstico presuntivo o confirmado de MCH, el 37 % lo hizo por algún síntoma y el 27 % por un control de rutina. Como se observa en la Figura 1, la disnea y el ángor fueron los síntomas más frecuentes, seguido de las palpitaciones y el sincope. El 41 % de los pacientes con disnea se encontraba en clase funcional III-IV, que representa el 9,5 % del total de pacientes.
En cuanto a los estudios diagnósticos, el 96 % de los pacientes tenía electrocardiograma (90 % ritmo sinusal, 91 % signos de hipertrofia ventricular izquierda), 96 % de los pacientes ecocardiograma, 71 % resonancia magnética cardíaca (RMC), 66 % ECG Holter de 24 horas. Se realizó test genético en el 33 % de los pacientes, cinecoronariografía en el 22 %, eco estrés con ejercicio en el 13 % y angiotomografía coronaria en el 7 % (Figura 2).
La localización de HVI, tanto en el ecocardiograma como en la RMC, se evidencio más frecuentemente en el septum interventricular (67 y 51 % respectivamente) y apical (9 y 13 % respectivamente). Se observó una diferencia significativa en la cuantificación de la fracción de eyección del ventrículo izquierdo (FEVI) por ecocardiografía en comparación con la RMC, con mediana (RIC 25-75) de 61 % (55-67 %) vs. 67 % (65- 74 %) respectivamente, p<0,001. Se observó gradiente obstructivo (>30 mmHg) del tracto de salida del ventrículo izquierdo (TSVI) en reposo en 35 pacientes (37 %) con una media de 43±26 mmHg en reposo y 62±39 mmHg en Valsalva (p<0,001).
Tabla 1. Datos basales
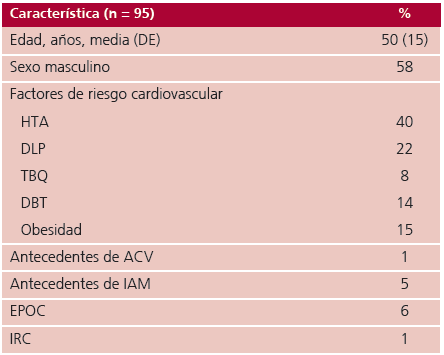
ACV: ataque cerebrovascular; DBT: diabetes mellitus; DE: desviación estándar DLP: dislipidemia; EPOC: enfermedad obstructiva crónica; HTA: hipertensión arterial; IAM: infarto agudo de miocardio; IRC: insuficiencia renal crónica; TBQ: tabaquismo
El test genético fue positivo en aproximadamente la mitad de los casos realizados (52 %); al momento de recabarse los datos de este registro, en uno de cada 3 pacientes estaba pendiente el resultado. Las alteraciones genéticas más frecuentemente identificadas fueron TNNT2 (5 pacientes), MYH7 (5 pacientes) y MYBPC3 (4 pacientes). Se interrogó acerca de los motivos por los que no se solicitó test genético y se reportó que en 19 casos no había cobertura por parte del plan de salud, en 16 casos no se disponía del test en su medio, en 12 casos los médicos nunca lo piden, 4 casos fueron rechazados por el paciente y 12 casos por otros motivos.
Se estudió a los familiares en el 44 % de los casos, mediante ecocardiograma (41 casos), RMC (15 casos), y test genético (14 casos).
Con respecto a la medicación, la mayoría usaron betabloqueantes (91 %), bloqueantes cálcicos (41 %), inhibidores/antagonistas del sistema renina angiotensina (IECA/ARAII 47 %), y estatinas (37 %). En la Figura 3 se describe la medicación que tenían los pacientes previamente a la consulta, y la agregada durante su seguimiento.
Si analizamos los tratamientos invasivos, de los 22 pacientes que consultaron con diagnostico confirmado de MCH o para segunda opinión, el 45 % tenían algún tratamiento invasivo previo: cardiodesfibrilador (CDI) 23 %, miomectomía 18 %, marcapasos 4 % y ablación con alcohol 4 %. Durante el seguimiento 22 pacientes requirieron tratamientos invasivos nuevos, CDI 18 %, marcapasos 3 %, resincronizador 1 % y miotomía septal 4 %.
Fig. 1. Síntomas de consulta (n = 95).
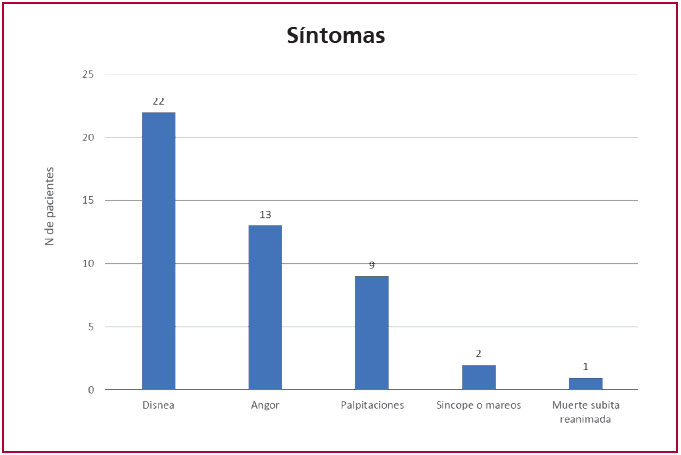
Fig. 2. Estudios realizados.
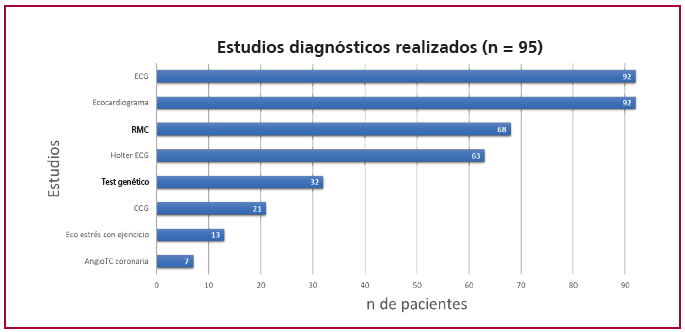
Angio TC: angiotomografía; CCG: cinecoronariografía; ECG: electrocardiograma; RMC: resonancia magnética cardíaca
Fig. 3. Medicación previa y agregada.
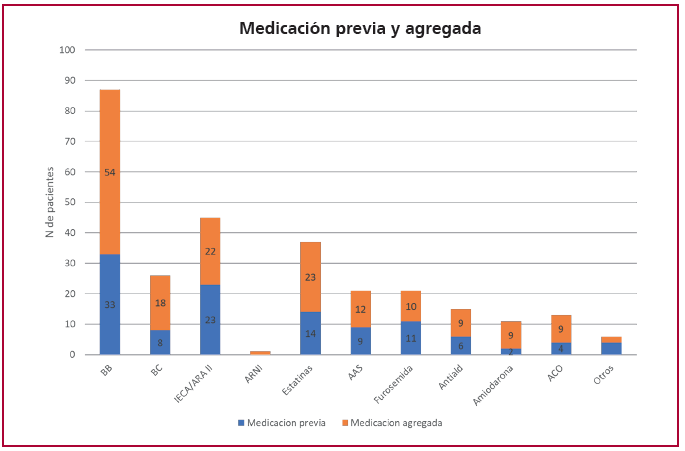
AAS; ácido acetilsalicílico; ACO: anticoagulación oral; Antiald: antialdosterónicos; ARA II: antagonistas de los receptores de angiotensina II; ARNI: inhibidores de neprilisina y receptores de angiotensina; BB: betabloqueantes; BC: bloqueantes cálcicos; IECA: inhibidores de la enzima convertidora de angiotensina
DISCUSIÓN
El presente estudio representa el primer acercamiento a la atención de pacientes con MCH en centros médicos no especializados en Argentina. Si bien la gran mayoría de cardiólogos que realizan el seguimiento de estos pacientes se concentra en Buenos Aires y CABA, existe representación de otras provincias: Santa Fe, Formosa, Catamarca, Tucumán, Río Negro, Mendoza y Chubut (ver apéndice)
En el campo de la MCH se han logrado avances, tanto en diagnóstico por imágenes como en tratamientos, que permiten a los pacientes una expectativa de vida similar a la población general. (11) Conocer los datos del mundo real en este tipo de patologías poco frecuentes, es importante para conocer el impacto de estas nuevas estrategias. Un ejemplo destacable es el Hypertrophic Cardiomyopathy Registry (HCMR), el mayor estudio prospectivo y multicéntrico, con 2755 pacientes de 44 centros de 6 países que además de datos clínicos, incluyó características de RMC, genotipificación y biomarcadores. (12-14) En nuestro medio no hay precedentes con respecto al registro de estos pacientes con foco en centros no especializados.
Dentro de los hallazgos más importantes de nuestro registro, se encontró que los pacientes se diagnosticaron a una edad similar a la observada en el HCMR, con una proporción similar de hipertensos, aunque con una mayor proporción de diabéticos. (12)
La disnea fue el síntoma predominante y un poco más de la cuarta parte de los pacientes fueron diagnosticados luego de una consulta rutinaria, por lo que se destaca el alto nivel de sospecha que se debe tener para el diagnóstico de esta patología ya que muchos se presentan asintomáticos u oligosintomáticos.
Una de las contribuciones más importantes que podemos mencionar del presente registro, es el referido a los estudios solicitados. El ECG y ecocardiograma son los más frecuentes, en probable relación con su amplia disponibilidad en el territorio. El ECG Holter fue utilizado en el 66 % de los casos, similar a lo reportado por el HMCR (60 %). Sin embargo, el eco estrés con ejercicio fue escasamente solicitado, aproximadamente en 1 de cada 10 pacientes, lo cual contrasta la sugerencia de la guía ESC 2023 de manejo de miocardiopatías, que lo propone como una indicación IB al momento de evaluar el gradiente en el TSVI. (5, 15-(17))
Si analizamos los datos ecocardiográficos, podemos observar que el porcentaje de pacientes con obstrucción del TSVI fue superior en nuestro registro en comparación con el HMCR (37 % vs. 18 %). Sin embargo, la media del gradiente en reposo fue menor en nuestro estudio (43 ± 26 mmHg vs. 69 ± 31 mmHg). Estas tendencias requieren una validación adicional mediante la inclusión de un mayor número de pacientes. Con respecto a la RMC, se puede resaltar que un porcentaje no menor de pacientes con diagnóstico altamente probable de MCH carece de dicho estudio (3 de cada 10). Este es otro dato que se contrapone con el HCMR en el cual 9 de cada 10 pacientes tienen el estudio realizado. (1218)
No obstante, los hallazgos en la RMC son similares, siendo la localización septal y apical las localizaciones más frecuentes y las medias de FEVI con valores semejantes (67 vs. 64 %). (12,13) Un aspecto interesante es la variabilidad en la cuantificación de la FEVI y la masa cardiaca por ecocardiografía y RMC, en probable relación con la resolución espacial de esta última. En lo que refiere al test genético, 2 de cada 3 pacientes no lo tienen realizado. El motivo más frecuente por el que el médico de seguimiento no lo solicita es la falta de medios o cobertura. (19)
En relación con el tratamiento farmacológico en nuestro registro el uso de betabloqueantes y bloqueantes cálcicos fue mayor con respecto al HCMR (91 vs. 57 % y 27 vs. 18,7 % respectivamente), lo cual podría deberse a la diferencia de pacientes con obstrucción del TSVI y en clase funcional III/IV (37 % vs. 18 % y 9,5 % vs. 7,2 %). La misma tendencia se observa en el uso de IECA/ARA II y estatinas (47 % vs. 23,7 % y 37 % vs. 27 %). Cabe destacar que el 46 % de los pacientes no presentaba tratamiento farmacológico previo a la consulta con el cardiólogo de seguimiento. (12)
Los tratamientos invasivos en la MCH son parte de los pilares en el manejo de estos pacientes, sobre todo en los que persisten sintomáticos a pesar del tratamiento farmacológico o tienen riesgo de muerte súbita. (5,(20)) En nuestro trabajo, 1 de cada 10 pacientes ingresados, ya tenían algún tratamiento previo a la primera consulta con el médico tratante, destacándose el implante de CDI. Una cuarta parte de los pacientes requirió terapia invasiva en el seguimiento, siendo predominante el implante de dispositivos. Resulta destacada la baja tasa de miomectomía y ablación con alcohol a pesar del porcentaje de pacientes de tipo obstructivo y disnea clase funcional III/IV.
En Argentina el sistema de salud se encuentra particularmente fragmentado, sumado a que existe inequidad de recursos entre las distintas provincias del territorio, lo cual podría explicar las diferencias que existen con otros países. Este punto nos podría ayudar a optimizar los recursos en este tipo de patología.
Como limitaciones podemos señalar que este registro cuenta con un tamaño muestral que no logra representatividad del territorio de la Argentina. Además, presenta debilidades inherentes a los registros retrospectivos y de participación voluntaria, que pueden generar sesgos de reporte.
CONCLUSIONES
El presente registro es el primer estudio de pacientes con MCH en Argentina que aporta datos del mundo real en cuanto a la atención de los centros no especializados. La disnea y el dolor precordial fueron los síntomas más frecuentes de presentación clínica. Se destacan al ECG y el ecocardiograma como los estudios pilares para la sospecha diagnóstica, hallándose en este sentido un espacio de mejora para el acceso a estudios más complejos. El tratamiento farmacológico es acorde a lo establecido en las guías internacionales.
Declaración de conflicto de intereses
Los autores declaran que no tienen conflicto de intereses.
(Véanse formularios de conflicto de intereses de los autores en la Web).
https://creativecommons.org/licenses/by-nc-sa/4.0/

©Revista Argentina de Cardiología
BIBLIOGRAFÍA
- Khorana AA, Francis CW, Culakova E, Kuderer NM, Lyman GH. Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapy. J Thromb Haemost. 2007;5:632-4. https://doi.org/10.1111/j.1538-7836.2007.02374.x
- Zou Y, Song L, Wang Z, Ma A, Liu T, Gu H, et al. Prevalence of idiopathic hypertrophic cardiomyopathy in China: a population-based echocardiographic analysis of 8080 adults. Am J Med. 2004;116:14-8. https://doi.org./10.1016/j.amjmed.2003.05.009
- Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, et al. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2022;79:372-89. https://doi.org/10.1016/j.jacc.2021.12.002
- Zipes DP, Libby P, Bonow RO, Mann DL, & Tomaselli GF. Braunwald. Tratado de cardiología: Texto de medicina cardiovascular. Elsevier Health Sciences (2019).
- Baxi AJ, Restrepo CS, Vargas D, Marmol-Velez A, Ocazionez D, Murillo H. Hypertrophic Cardiomyopathy from A to Z: Genetics, Pathophysiology, Imaging, and Management. RadioGraphics 2016;36:335–54. https://doi.org/10.1148/rg.2016150137
- Arbelo E, Protonotarios A, Gimeno JR, Arbustini E, Barriales- Villa R, Basso C, et al; ESC Scientific Document Group. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023;44:3503-626. https://doi.org/10.1093/eurheartj/ehad194
- Maron BJ, Rowin EJ, Udelson JE, Maron MS. Clinical Spectrum and Management of Heart Failure in Hypertrophic Cardiomyopathy. JACC: Heart Failure 2018;6:353–63. https://doi.org/10.1016/j.jchf.2017.09.011
- Rowin EJ, Maron BJ, Maron MS. The Hypertrophic Cardiomyopathy Phenotype Viewed Through the Prism of Multimodality Imaging: Clinical and Etiologic Implications. JACC Cardiovasc Imaging. 2020;13:2002-16. https://doi.org/10.1016/j.jcmg.2019.09.020
- Fernández A, Acunzo RS, Avegliano G, Casabé JH, Dumont CA, Hita A, Ortiz M, Pérez de Arenaza D, y cols. Consenso Argentino de diagnóstico y tratamiento de la Miocardiopatía Hipertrófica 2016. Sociedad Argentina de Cardiología. Rev Argent Cardiol 2017;85(Suplemento 2):1-78.
- Kotkar KD, Said SM, Dearani JA, Schaff HVl. “Hypertrophic obstructive cardiomyopathy: the Mayo Clinic experience.” Annals of Cardiothoracic Surgery 2017:6:329-36. https://doi.org/10.21037%2Facs.2017.07.03
- Hodges K, Rivas CG, Aguilera J, Borden R, Alashi A, Blackstone EH, et al. “Surgical management of left ventricular outflow tract obstruction in a specialized hypertrophic obstructive cardiomyopathy center.” The Journal of Thoracic and Cardiovascular Surgery 2019:157:2289–99. https://doi.org/10.1016/j.jtcvs.2018.11.148
- Maron, B. J., Rowin, E. J., Casey, S. A., & Maron, M. S. How hypertrophic cardiomyopathy became a contemporary treatable genetic disease with low mortality: shaped by 50 years of clinical research and practice. JAMA cardiology 2016;1:98-105. https://doi.org/10.1001/jamacardio.2015.0354
- Neubauer S, Kolm P, Ho CY, Kwong RY, Desai MY, Dolman Sf, et al; HCMR Investigators. Distinct Subgroups in Hypertrophic Cardiomyopathy in the NHLBI HCM Registry. J Am Coll Cardiol. 2019;74:2333-45. https://doi.org/10.1016/j.jacc.2019.08.1057
- Wigle ED, Sasson Z, Henderson MA, Ruddy TD, Fulop J, Rakowski H, et al. Hypertrophic cardiomyopathy. The importance of the site and the extent of hypertrophy. A review. Progress in Cardiovascular Diseases .1985;28(1):1-83. https://doi.org/10.1016/0033-0620(85)90024-6
- Kramer CM, Appelbaum E, Desai MY, Desvigne-Nickens P, DiMarco JP, Friedrich MG, et al. Hypertrophic Cardiomyopathy Registry: The rationale and design of an international, observational study of hypertrophic cardiomyopathy. Am Heart J. 2015;170:223-30. https://doi.org/10.1016/j.ahj.2015.05.013
- Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2020;76:159-240. https://doi.org/10.1016/j.jacc.2020.08.045
- Shah JS, Esteban MT, Thaman R, Sharma R, Mist B, Pantazis A, et al. Prevalence of exercise-induced left ventricular outflow tract obstruction in symptomatic patients with non-obstructive hypertrophic cardiomyopathy. Heart 2008;94:1288–1294. https://doi.org/10.1136/hrt.2007.126003
- Maron MS, Olivotto I, Zenovich AG, Link MS, Pandian NG, Kuvin JT, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation CIRCULATIONAHA.106.644682
- Rudolph A, Abdel-Aty H, Bohl S, Boye P, Zagrosek A, Dietz R, et al. Noninvasive detection of fibrosis applying contrast-enhanced cardiac magnetic resonance in different forms of left ventricular hypertrophy relation to remodeling. J Am Coll Cardiol 2009;53: 284-291. https://doi.org/10.1016/j.jacc.2008.08.064
- Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003;107:2227–2232. https://doi.org/10.1161/01.CIR.0000066323.15244.5419. Maron BJ, Spirito P, Shen WK, Haas TS, Formisano F, Link MS, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA 2007;298:405–412. https://doi.org/10.1001/jama.298.4.405
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996;111: 586-594. https://doi.org/10.1016/S0022-5223(96)70310-0
