
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is the most common genetic heart disease, with an estimated prevalence between 1 in 200 and 500 people. (1,2) Its etiology is related to mutations in genes encoding sarcomere proteins, resulting in center ventricular hypertrophy (LVH), myofibrillar disarray, and progressive myocardial fibrosis. (3,4)
The diagnosis remains a significant challenge, as increased wall thickness and chamber dilation are not exclusive to HCM but can also be observed in infiltrative cardiomyopathies that present as phenocopies. (5-7)
Current treatments include pharmacological strategies and invasive options, such as septal surgery and device implantation, which have been shown to improve survival and quality of life. (8-10) However, there are few centers specialized in this disease, so most patients are followed up at institutions not specifically dedicated to HCM.
This study presents the final results of the Hypertrophic Cardiomyopathy Registry in Non-specialized Centers, with the aim of providing data to assess the limitations in access to and use of advanced diagnostic methods outside of referral centers.
METHODS
We conducted a nationwide, observational, retrospective, and multicenter study in patients with confirmed or highly probable diagnosis of HCM.
The participants were clinical cardiologists practicing in outpatient clinics. For the purposes of this study, a nonspecialized center was defined as one without a structured program dedicated to managing HCM or a systematic, multidisciplinary approach. These centers also had non-systematic access to advanced septal reduction therapies and high-complexity tests, such as cardiac magnetic resonance imaging or genetic testing.
Hypertrophic cardiomyopathy was defined as the presence of center ventricular hypertrophy with a maximum wall thickness >15 mm in a non-dilated center ventricle, documented by echocardiography and/or cardiac magnetic resonance imaging, in the absence of other conditions resulting in such hypertrophy. (2-6)
Cases that met the morphological and imaging criteria and had a positive genetic test result were categorized as confirmed HCM. (2,4,8-10) Patients who met all clinical and imaging criteria, but had not undergone genetic testing, were considered to be highly probable cases of MCH.
In all cases, the final diagnostic adjudication was center to the discretion of the treating cardiologist, without centralized review of images or validation of diagnoses by an independent reviewer.
Patients with conditions associated with secondary ventricular hypertrophy as the sole mechanism (such as infiltrative cardiomyopathies, hypertension, valvular heart disease, and high-performance athletes, among others) were excluded, as were those with HCM who were being followed up at specialized centers.
Data were collected on the REDCap platform of the Argentine Society of Cardiology between June 1, 2023, and September 1, 2024.
Statistical analysis
Qualitative variables were expressed as absolute and relative frequencies. Continuous variables were described as mean and standard deviation (SD), or median and interquartile range (IQR 25-75), according to their distribution.
Because of the descriptive nature of the study, the analysis focused primarily on presenting descriptive statistics. Categorical variables were compared using the chi-square test or the Fisher's exact test, as appropriate. For continuous variables, the Student's t test or the Mann-Whitney test were used to compare independent groups, and the Wilcoxon signed-rank test was used to assess changes in paired data, as appropriate.
A p-value < 0.05 was considered statistically significant. All the statistical calculations were performed using R and Phython software packages.
Ethical considerations
The study protocol was reviewed and approved by the institutional review board of Hospital General de Agudos Donación F. Santojanni of the City of Buenos Aires. The research was conducted in accordance with the ethical principles established in the Declaration of Helsinki (11) and its subsequent amendments, as well as with current local regulations regarding clinical research.
RESULTS
A total of 160 patients from 8 provinces in Argentina (Buenos Aires, Santa Fe, Formosa, Catamarca, Tucuman, Rio Negro, Mendoza, and Chubut) were included. Mean age was 48 years and 60.6% were men. The most common cardiovascular risk factors were hypertension (46.7%), dyslipidemia (31.4%), obesity (18.2%), diabetes (18.2%), and tobacco use (11.9%). The prevalence of comorbidities associated with cardiovascular disease was low: chronic obstructive pulmonary disease (4.4%), prior myocardial infarction (3.8%), chronic kidney disease (2.2%), anemia (2.5%), and stroke (1.2%) (Table 1). Among patients with a family history of sudden cardiac death, 66.7% corresponded to events occurring in individuals <45 years of age.
Table 1
Baseline data
| Characteristic (n=160) | ||
|---|---|---|
| Age, years, mean ± SD | 48±16 | |
| Gender (%) | ||
| Male | 60.6 | |
| Female | 39.4 | |
| CVRF (%) | ||
| HTN | 46.7 | |
| DLP | 31.4 | |
| TU | 11.9 | |
| DBT | 18.2 | |
| Obesity | 18.2 | |
| History of sudden death (%) | 17.5 | |
| < 45 years | 66.7 | |
| Comorbidities(%) | ||
| Prior AMI | 3.8 | |
| COPD | 4.4 | |
| CKD | 2.2 | |
| Anemia | 2.5 | |
| Prior stroke | 1.2 | |
AMI: acute myocardial infarction; CKD: chronic kidney disease; COPD: chronic obstructive pulmonary disease; CVRF: cardiovascular risk factors; DBT: diabetes; HTN: hypertension; SD: standard deviation; TU: tobacco use
The most common reason for consultation was a presumptive or confirmed diagnosis of HCM (43.4%), followed by symptoms (28.3%) and routine checkups (28.3%) (Figure 1). Among symptomatic patients, the most common symptoms were dyspnea (n=30) and angina (n=15), followed by palpitations (n=11), syncope (n=3), and one case of resuscitated sudden cardiac death. Among patients presenting with dyspnea, 22.7% were in New York Heart Association (NYHA) functional class (FC) I, 40.9% in FC II, and 36.4% in FC III-IV.
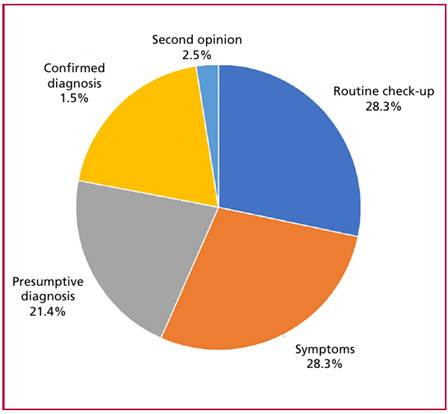
Median time from the diagnosis of HCM to inclusion in the registry was 3.9 years (IQR 2.0-10.0). An analysis of the frequency of follow-up visits revealed that 64.8% of patients had attended the outpatient clinic within the past 6 months, 23.9% between 6 and 12 months, and 11.4% more than 12 months ago.
Regarding ancillary tests, an electrocardiogram was performed in all patients; 88.5% were in sinus rhythm, and 87.5% showed signs of center ventricular hypertrophy. Sixty percent of the cohort underwent cardiac magnetic resonance imaging (CMRI) and 49.3% 24-hour Holter monitoring. In addition, genetic testing was performed in 40% of the patients, coronary angiography or coronary computed tomography angiography in 20%, and stress echocardiography in 10.6% (Figure 2).
Fig. 2
Ancillary tests during follow-up
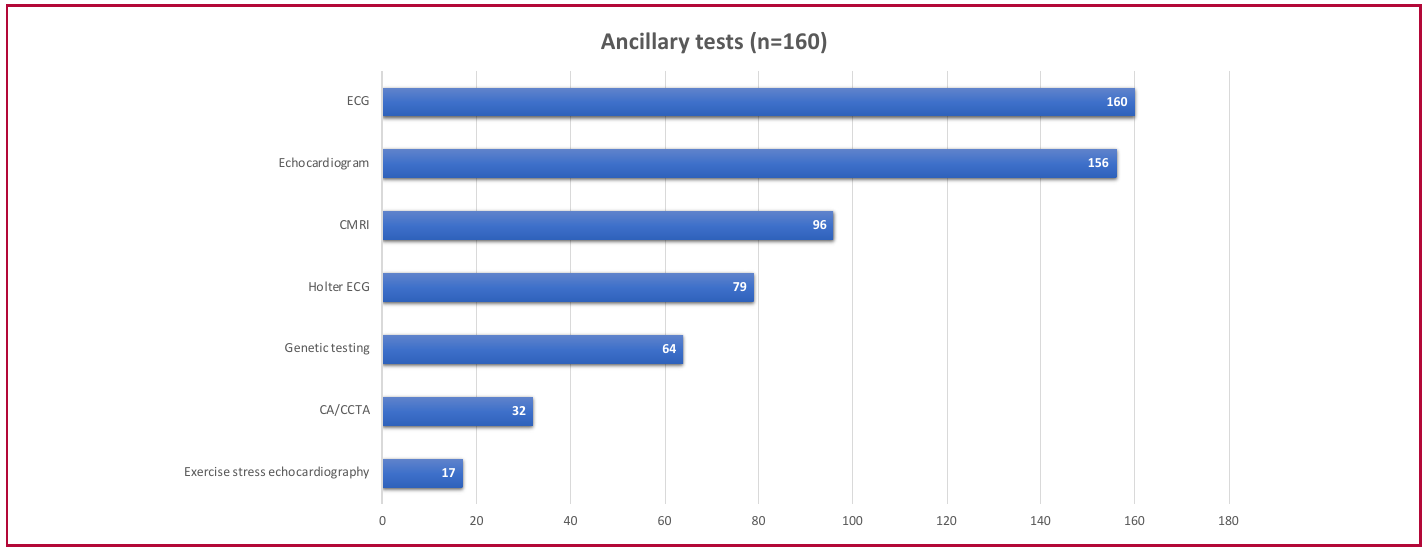
CA: coronary angiography; CCTA: coronary computed tomography angiography; CMRI: magnetic resonance imaging; ECG: electrocardiogram
Hypertrophy was predominantly septal (84.4% on echocardiography and 78.9% on CMRI) and apical (11.2% and 15%, respectively). There were no significant differences in center ventricular ejection fraction measured by echocardiography or CMRI [median 61% (IQR 52-70) vs. 66% (IQR 55-77); p = 0.114].
Among the 96 patients evaluated with CMRI, 81.2% had late gadolinium enhancement, predominantly with an intramyocardial pattern (92%). This pattern was most common in the interventricular septum (75%) with a patchy distribution in 55.4%; median fibrosis score was 5.1% (IQR 2.8-17).
Left ventricular outflow tract obstruction (>30 mmHg) was observed in 55 patients (34.3%). Median resting gradient was 43 mmHg (IQR 28-66) and increased to 62 mmHg (IQR 37-82) during the Valsalva maneuver.
In patients undergoing genetic testing, 82.8% tested positive for pathogenic variants or likely pathogenic variants (n=53/64). The most common variants were identified in the sarcomere genes MYH7 (n=21), MYBPC3 (n=13), and TNNT2 (n=6).
Patients who underwent genetic testing were compared with those without genetic testing (Table 2). Patients who underwent genetic testing were significantly younger [median age 37 years (IQR 25-48) vs. 58 years (QR 45-70); p < 0.001] and had a higher prevalence of a family history of sudden cardiac death (25% vs. 12.5%; p = 0.043). The sudden cardiac risk score was similar in both groups.
| Variable | Genetic testing n=64 | No genetic testing n=96 | p |
|---|---|---|---|
| Age, years (median, IQR) | 37 (25-48) | 58 (45-70) | <0 .001 |
| Male, n (%) | 38 (59.3) | 58 (60.4) | 0 .401 |
| Family history of SCD, n (%) | 16 (25) | 12 (12.5) | 0 .038 |
| Dyspnea, n (%) | 12 (18.7) | 18 (18.7) | 1 |
| Angina, n (%) | 7 (10.9) | 8 (8.3) | 0 .592 |
| LVOT obstruction, n (%) | 15 (23.4) | 21 (21.8) | 0 .818 |
| CMRI, n (%) | 33 (51.5) | 63 (65.6) | 0 .564 |
| Fibrosis on CMRI, n (%) | 26/31 (83.8) | 52/62 (83.8) | 0 .679 |
| ESC SCD risk score, % (median, IQR) | 5.8 (3.2-8.1) | 6.5 (3.5-9.0) | 0 .417 |
CMRI: cardiac magnetic resonance imaging; ESC: European Society of Cardiology; IQR: interquartile range; LVOT: center ventricular outflow tract; SCD: sudden cardiac death.
The main reasons for not performing genetic testing were lack of health insurance (n=29), local unavailability (n=16), and the treating physician’s decision (n=15). Other reasons were reported in a smaller group (n=21), while the cause could not be determined in the remaining patients.
Family screening was performed in 44.9% of the cohort (n=72), primarily through echocardiography (n=59), followed by genetic testing (n=35) and cardiac magnetic resonance imaging (n=22).
The most common medications prescribed were beta-blockers (42.5%), renin-angiotensin system inhibitors (16.9%), calcium channel blockers (15.6%), and furosemide (11.2%). Figure 3 shows the medications patients were receiving prior to their first visit and those subsequently prescribed based on clinical criteria.
Fig. 3
Medications before and after the first visit
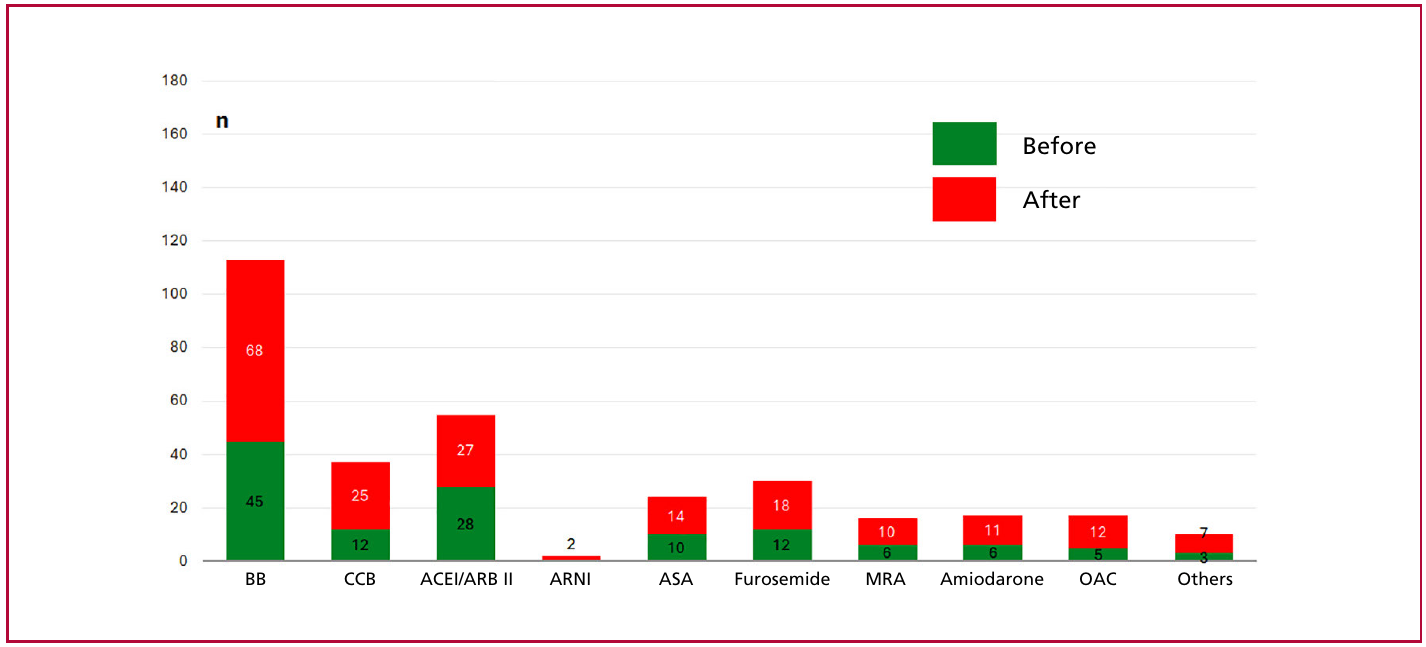
ACEI: angiotensin-converting enzyme inhibitor; ARB II: angiotensin II receptor antagonist; ARNI: angiotensin receptor/neprilysin inhibitor; ASA: acetylsalicylic acid; BB: beta-blocker; CCB: calcium channel blocker; MRA: mineralocorticoid receptor antagonist; OAC: oral anticoagulant.
Among the 20 patients with a prior diagnosis of HCM who were being followed up by their treating cardiologists, 11 had an implantable cardioverter-defibrillator (ICD) implanted, 4 had undergone myectomy, 3 had pacemakers, and 2 had undergone alcohol septal ablation. In the entire cohort analyzed, 35 patients had a history of therapeutic interventions following the diagnosis of HCM, including 26 ICD implants, 4 pacemaker implants, 4 septal myectomy procedures, and 1 resynchronization device implant. Myectomy significantly reduced the LVOT gradient in two-thirds of the cases.
Of the 26 patients who received an ICD during follow-up, the European Society of Cardiology (ESC) score was available in 10 cases and the American Heart Association (AHA) score in 15. Median ESC score was 6.5% (IQR 5.1-8.2). According to the risk categories,
60% (6 patients) were in the high-risk category (score ≥ 6%), 2 patients were in the intermediate-risk category (4-6%), and 2 were in the low-risk category (<4%). Median AHA score was 6.3 (IQR 3.2-9.4). Three appropriate shocks (11.5% of patients with an ICD) were documented at the time of the registry.
DISCUSSION
This registry constitutes the first national study analyzing patients with HCM treated in non-specialized centers. Its relevance lies in the fact that most international literature comes from referral centers, where diagnosis and management are carried out following standardized protocols and with broad access to advanced imaging tests. (12-16) Conversely, our results reflect actual clinical practices in a country with significant differences in the availability of resources and access to specialized care.
While most of the cardiologists who monitored these patients practice in Buenos Aires and the City of Buenos Aires (CABA), centers from several inland provinces (Santa Fe, Formosa, Catamarca, Tucumán, Río Negro, Mendoza, and Chubut) were also included.
The diagnosis of HCM remains a challenge, even in settings with advanced resources, due to its heterogeneous clinical presentation and overlap with phenocopies. In our registry, the diagnosis was center to the discretion of the treating cardiologist and was not always supported by highly complex ancillary tests, reflecting the status of real-world clinical practice in non-specialized centers. This characteristic, which could be regarded as a limitation, also contributes to the study, as it facilitates the identification of diagnostic challenges in routine care settings outside of referral centers.
The mean age in our cohort (48 years) was similar to that reported in international registries, such as the Hypertrophic Cardiomyopathy Registry (HCMR, 49 years),(17) the EURObservational Registry (47 years), (18) the Portuguese Registry (53 years), (19) the Italian (44 years), (20) and the Sarcomeric Human Cardiomyopathy Registry (SHaRe, 45.8 years).(21) Likewise, we observed a male predominance (60.6%), comparable to that described in the HCMR (71%) and SHaRe (63%). However, there is a growing consensus that the underrepresentation of women suggests underdiagnosis rather than a true difference in disease incidence.
A family history of sudden cardiac death was present in 17.5% of our patients, a proportion similar to that reported in the SHaRe registry. The systematic assessment of this variable is a key aspect of our registry, due to its impact on risk stratification in HCM.
Regarding the reasons for diagnosis, 28.3% of cases involved symptomatic patients, and 28.3% were detected during routine checkups. These proportions are similar to those reported in the EURObservational and Portuguese registries. However, in our cohort 36.4% of patients with dyspnea were in FC III-IV, whereas in most international registries this proportion was considerably lower (<10%), with a clear predominance of patients in FC I-II (≈80%). This difference may reflect a later diagnosis in our setting, likely associated with limitations in early access to specialists and advanced diagnostic methods. (9,10)
Hypertrophy was predominantly septal and, to a lesser extent, apical, consistent with findings described in the international literature. Similarly, the prevalence of fibrosis detected by CMRI was high (81.2% among the evaluated patients), consistent with previous registry reports. (7) However, this finding should be interpreted in the context of a selected population since CMRI was not systematically performed across the entire cohort, but rather in a subgroup of patients with clinical indications. This may have overestimated the prevalence of fibrosis.
In contrast, the proportion of patients undergoing stress echocardiography was markedly low (10.6%), despite this tool being accessible and essential for differentiating between baseline and induced obstruction through the Valsalva maneuver or during exercise. This finding likely reflects the absence of standardized protocols and the need for greater integration of stress echocardiography into routine diagnostic pathways at non-specialized centers.
Genetic testing was performed in 40% of patients, a proportion comparable to that of the Portuguese registry (51%) and somewhat lower than that of the SHaRe (60%).(19,21) The diagnostic performance was high (82.8%), with a predominance of classic sarcomere variants (MYH7, MYBPC3, and TNNT2). This finding should be interpreted in the context of selection bias. In the comparative analysis, patients undergoing genetic testing were significantly younger [median age 37 years (IQR 25-48) vs. 58 years (QR 45-70); p < 0.001] and had a higher prevalence of a family history of sudden cardiac death (25% vs. 12.5%; p = 0.043), with no differences observed in other clinical or morphological variables. These findings suggest that the test was primarily indicated in individuals with a higher suspicion of a genetic etiology rather than based on clinical severity or structural phenotype. When considered as a whole, this explains the higher diagnostic performance compared to that reported in unselected cohorts. Additionally, it suggests that the differences observed in the use of various diagnostic methods are more attributable to variations in their implementation in clinical practice than to their actual availability. Consequently, there is an opportunity to enhance the standardization of the diagnostic approach in non-specialized centers.
Likewise, the primary barrier to genetic testing was the lack of insurance coverage, which restricted not only individual access but also the implementation of family cascade screening strategies. In this context, only 44.9% of relatives were evaluated, predominantly through echocardiography, which contrasts with reports from European and American registries. (17-21)
Beta-blockers were the drugs most commonly used, consistent with reports from other registries. In the cohort analyzed, 26 patients had an implantable cardioverter-defibrillator (ICD) implanted due to disease progression.
The median ESC sudden cardiac death risk score was 6.5%, and most patients with an implanted ICD were in the high-risk category (≥6%). These findings are consistent with the recommendations of the ESC guidelines for ICD implantation in primary prevention. (5)
However, ICDs had been also implanted in low or intermediate-risk patients, which likely reflects the consideration of additional clinical variables, such as syncope, non-sustained ventricular tachycardia, late gadolinium enhancement on CMRI, or family history in the decision-making process. The lack of stratification scores in all cases made it impossible to perform a more comprehensive analysis. However, the overall trend suggests that most ICDs were implanted in high-risk patients.
These findings should be interpreted in the context of a cohort with a prolonged clinical course since diagnosis, a median of nearly 4 years, and a wide range of follow-up visits. This aspect could influence the frequency of the observed interventions, including ICD implantation, as it reflects different stages of disease progression rather than events occurring within a defined follow-up period.
The rate of myectomy procedures was low (4 cases on admission and 4 during follow-up), which is likely related to the lack of centers with expertise in interventional procedures in the country.
Our findings underscore two key challenges in the management of HCM in Argentina. First, inequity in access to diagnostic tests. Although stress echocardiography is widely available across most centers, its underutilization reflects the absence of standardized protocols and the need for greater integration into routine diagnostic pathways. In contrast, genetic testing was performed at a relatively high rate despite being less accessible, although this was limited by health system coverage.
Second, the limited dissemination and implementation of guidelines, recommendations, and diagnostic algorithms. The heterogeneous use of fundamental tools underscores the necessity to enhance training and integrate standardized diagnostic approaches in non-specialized centers. This could help reduce late diagnoses and the higher proportion of patients in FC III-IV observed in our registry.
In this context, the differences observed reflect not only the structural heterogeneity of the Argentine healthcare system but also the inequities in care pathways, including referral, timely diagnosis, and the implementation of risk stratification strategies.
Based on these findings, specific courses of action have been identified focused on improving care:
- Development of updated national guidelines adapted to the Argentine context considering the available resources and defining practical algorithms for diagnosis and treatment. In this regard, we can only welcome the recent publication of the Argentine Consensus on the Diagnosis and Treatment of Hypertrophic Cardiomyopathy, which provides a comprehensive review of the topic and recommendations aligned with scientific advances and local realities. (22)
Implementation of continuing medical education programs aimed at professionals in non-specialized centers.
Creation of referral and counter-referral networks to facilitate the timely referral of patients requiring advanced interventions.
Promotion of equity strategies for the access to genetic testing and family screening.
When considered as a whole, our results, which are consistent with large international registries in terms of clinical characteristics, underscore the significant differences in the implementation of diagnostic and therapeutic processes. They also provide insight into the management of HCM in real-world clinical practice and set the groundwork for developing local strategies and policies that bridge the gap between the available evidence and its implementation in daily practice.
Study limitations
The study has several limitations that should be considered to interpret the results. First, the sample size and geographic distribution of the participating centers do not allow extrapolating the findings to the entire national population. Second, the retrospective nature of the registry and voluntary participation introduce potential selection and reporting biases, with possible over-representation of more complex patients or with tighter follow-up.
Likewise, the heterogeneous availability and use of ancillary tests across centers and provinces affect internal comparability and limit the standardization of the diagnostic approach. In this context, the final diagnosis of HCM was center to the discretion of the treating cardiologist which reflects actual clinical practice, although it may have introduced variability in case definition.
The lack of a systematic prospective follow-up limited the assessment of clinical events over time. Therefore, the findings are interpreted primarily in terms of the patients’ history at the time of enrollment. Finally, the incomplete availability of key studies-such as stress echocardiography, CMRI, genetic testing, and risk stratification scores-restricted the comprehensive analysis of some subgroups and may have influenced the interpretation of certain results.
CONCLUSIONS
This first Argentine registry of patients with HCM treated in non-specialized centers allowed for a comprehensive characterization of the population and the diagnostic and therapeutic strategies used in realworld clinical practice. Although our findings are similar to those of large international registries in terms of clinical characteristics, we identified significant differences in the use of ancillary tests, timely diagnosis, and implementation of therapeutic strategies. These findings underscore the presence of inequities in the access to diagnostic resources and care pathways, including referral, risk stratification, and family-centered care. In this context, the results of the present study provide local evidence to guide the development of concrete strategies, including healthcare team training, the consolidation of referral networks, and the adaptation of international guidelines to the Argentine context, with the goal of optimizing the diagnosis and management of this disease nationwide.
Conflicts of interest None declared.
(See authors' conflict of interests forms on the web).
Acknowledgments
The authors thank the researchers participating in the registry and the Argentine Society of Cardiology for the logistical and academic support.
Funding
The authors declare that this study did not receive any public, private, or institutional funding.