
INTRODUCCIÓN
La miocardiopatía hipertrófica (MCH) constituye la enfermedad genética cardiovascular más frecuente, con una prevalencia estimada de 1 en 200 a 500 individuos. (1,2) Su etiología se relaciona con mutaciones en genes que codifican proteínas del sarcómero, lo que conduce al desarrollo de hipertrofia ventricular izquierda (HVI), desorganización miofibrilar y fibrosis miocárdica progresiva. (3,4)
El diagnóstico continúa representando un verdadero desafío, ya que el aumento del espesor parietal y la dilatación de cavidades no son exclusivos de la MCH, sino que también pueden observarse en miocardiopatías infiltrativas que actúan como fenocopias. (5-7)
Los tratamientos actuales incluyen estrategias farmacológicas y opciones invasivas, como la cirugía septal y el implante de dispositivos, que han demostrado mejorar la sobrevida y la calidad de vida. (8-10) Sin embargo, son escasos los centros especializados en esta patología, por lo que la mayoría de los pacientes son seguidos en instituciones no dedicadas especialmente a la misma.
Este estudio presenta los resultados definitivos del Registro de Miocardiopatía Hipertrófica en centros no especializados, con el objetivo de aportar datos que permitan dimensionar las limitaciones en el acceso y la utilización de métodos diagnósticos avanzados fuera de los centros de referencia.
MATERIAL Y MÉTODOS
Se realizó un estudio observacional, retrospectivo y multicéntrico de alcance nacional en pacientes con MCH confirmada o altamente probable.
Participaron cardiólogos clínicos que desarrollan su actividad asistencial en consultorios ambulatorios. A los fines del estudio, se consideraron centros no especializados aquellos sin un programa estructurado dedicado al manejo de la MCH, sin un abordaje multidisciplinario sistemático y con acceso no sistemático a terapias avanzadas de reducción septal y a estudios de alta complejidad, como resonancia magnética cardíaca o test genético.
Se consideró miocardiopatía hipertrófica a la presencia de HVI con espesor parietal máximo >15 mm, en un ventrículo izquierdo no dilatado, documentada por ecocardiografía y/o resonancia magnética cardíaca, en ausencia de otras condiciones capaces de justificar ese grado de hipertrofia. (2-6)
Se clasificó como MCH confirmada a los casos que, además de cumplir los criterios morfológicos e imagenológicos, contaban con test genético compatible. (2,4,8-10) Se consideró MCH altamente probable a aquellos pacientes que cumplían criterios clínico-imagenológicos completos, pero en quienes no se había realizado estudio genético. En todos los casos, la adjudicación diagnóstica final quedó a criterio del cardiólogo tratante y no se realizó adjudicación central de imágenes ni validación independiente de los diagnósticos.
Se excluyeron pacientes con condiciones capaces de generar hipertrofia ventricular secundaria como único mecanismo (miocardiopatías infiltrativas, hipertensión arterial, valvulopatías, entrenamiento deportivo de alto rendimiento, entre otras) y aquellos con MCH en seguimiento en centros especializados.
La recolección de datos se efectuó a través de la plataforma REDCap de la Sociedad Argentina de Cardiología, entre el 1 de junio de 2023 y el 1 de septiembre de 2024.
Análisis estadístico
Las variables cualitativas se expresaron como frecuencias absolutas y relativas. Las variables cuantitativas se describieron mediante media ± desviación estándar (DE) o mediana y rango intercuartílico (RIC 25-75), según la distribución de los datos.
Dado el carácter descriptivo del estudio, el análisis se centró principalmente en la presentación de estadísticas descriptivas. En los casos en que se realizaron comparaciones exploratorias, se utilizaron pruebas de chi cuadrado o exacta de Fisher para variables categóricas.Las variables continuas se analizaron mediante prueba t de Student o prueba de Mann-Whitney para comparaciones entre grupos independientes, y mediante prueba de rangos con signo de Wilcoxon para comparaciones apareadas, según correspondiera.
Se consideró estadísticamente significativo un valor de p < 0,05. Todos los análisis se realizaron utilizando R y Python.
Consideraciones éticas
El protocolo del estudio fue evaluado y aprobado por el Comité de Ética en Investigación del Hospital General de Agudos Donación F. Santojanni de la Ciudad de Buenos Aires. La investigación se condujo de acuerdo con los principios éticos establecidos en la Declaración de Helsinki (11) y sus enmiendas posteriores, así como con las normativas locales vigentes en materia de investigación clínica.
RESULTADOS
Se incluyeron 160 pacientes provenientes de 8 provincias de Argentina (Buenos Aires, Santa Fe, Formosa, Catamarca, Tucumán, Río Negro, Mendoza y Chubut). La edad media fue de 48 años y predominó el sexo masculino (60,6 %). Los factores de riesgo cardiovascular más frecuentes fueron hipertensión arterial (46,7 %), dislipidemia (31,4 %), obesidad (18,2 %), diabetes (18,2 %) y tabaquismo (11,9 %). La prevalencia de comorbilidades con asociación cardiovascular fue baja: enfermedad pulmonar obstructiva crónica (4,4 %), infarto de miocardio previo (3,8 %), insuficiencia renal crónica (2,2 %), anemia (2,5 %) y accidente cerebrovascular (1,2 %) (Tabla 1). Entre los pacientes con antecedentes familiares de muerte súbita, el 66,7 % correspondió a eventos ocurridos en individuos <45 años.
Tabla 1
Datos basales
| Característica (n=160) | |
|---|---|
| Edad, años, media ± DE | 48±16 |
| Sexo (%) | |
| Masculino | 60,6 |
| Femenino | 39,4 |
| FRCV (%) HTA | 46,7 |
| DLP | 31,4 |
| TBQ | 11,9 |
| DBT | 18,2 |
| Obesidad | 18,2 |
| Antecedente de muerte súbita (%) | 17,5 |
| Menos de 45 años | 66,7 |
| Comorbilidades (%) | |
| IAM previo | 3,8 |
| EPOC | 4,4 |
| IRC | 2,2 |
| Anemia | 2,5 |
| ACV previo | 1,2 |
ACV: accidente cerebrovascular; DBT: diabetes; DE: desvío estándar; DLP: dislipidemia; EPOC: enfermedad pulmonar obstructiva crónica; FRCV: factores de riesgo cardiovascular; HTA: hipertensión arterial; IAM: infarto agudo de miocardio; IRC: insuficiencia renal crónica; TBQ: tabaquismo
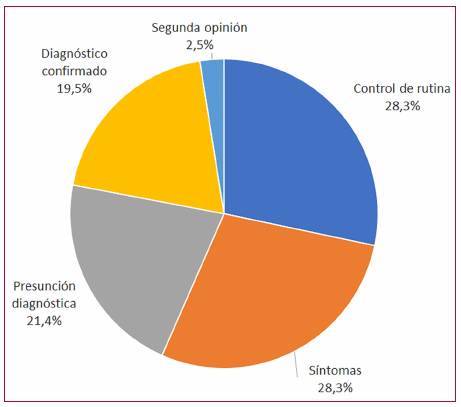
El motivo de consulta más frecuente fue el diagnóstico presuntivo o confirmado de MCH (43,4 %), seguido por síntomas (28,3 %) y controles de rutina (28,3 %) (Figura 1). Entre los pacientes sintomáticos, los síntomas más frecuentes fueron disnea (n=30) y angina (n=15), seguidos de palpitaciones (n=11), síncope (n=3) y un caso de muerte súbita reanimada. Entre los pacientes que presentaron disnea, el 22,7 % se encontraba en clase funcional (CF) I según la New York Heart Association (NYHA), el 40,9 % en CF II y el 36,4 % en CF III-IV.
El tiempo desde el diagnóstico de MCH hasta el momento de inclusión en el registro mostró una mediana de 3,9 años (RIC 2,0-10,0). Respecto a la recencia del seguimiento, el 64,8 % de los pacientes había tenido una consulta en los últimos 6 meses, el 23,9 % entre 6 y 12 meses y el 11,4 % más allá de 12 meses.
En cuanto a los estudios complementarios, todos los pacientes tuvieron electrocardiograma -con ritmo sinusal en el 88,5 % y signos de hipertrofia ventricular izquierda en el 87,5 %- y el 97,5 % fue evaluado mediante ecocardiograma. La resonancia magnética cardíaca (RMC) se realizó en el 60 % de la cohorte y el monitoreo Holter de 24 horas en el 49,3 %. Además, se efectuaron estudios genéticos en el 40 % de los pacientes, cinecoronariografía o angiotomografía coronaria en el 20 %, y ecocardiograma de estrés en el 10,6 % de la cohorte (Figura 2).
Fig. 2
Estudios realizados en el seguimiento
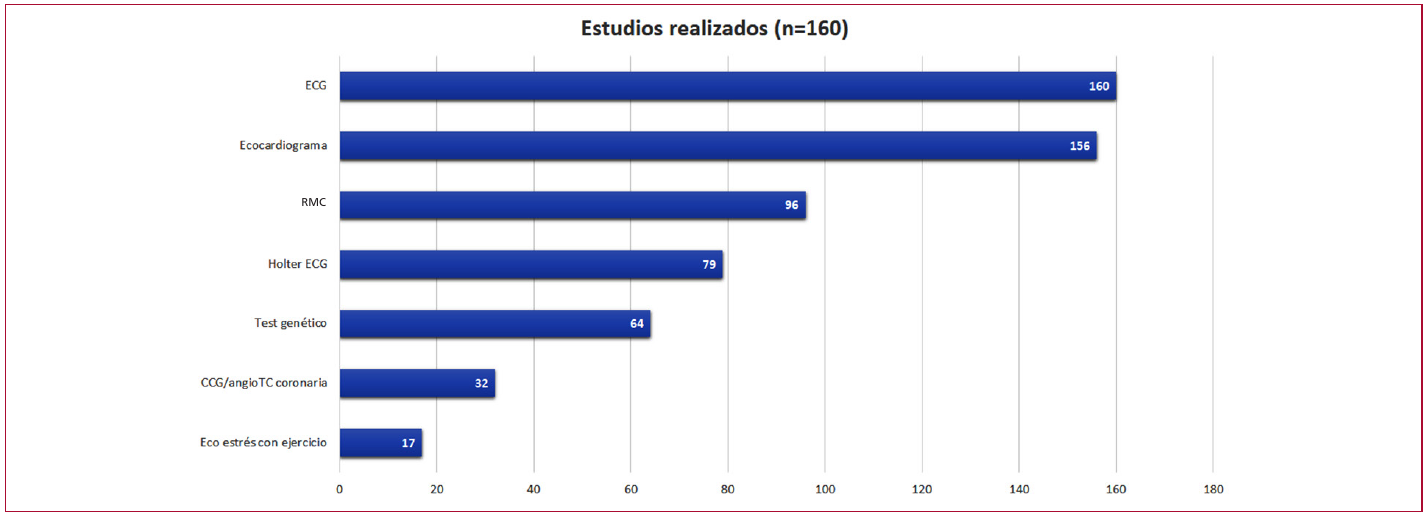
angioTC: angiotomografía; CCG: cinecoronariografía; ECG: electrocardiograma; RMC: resonancia magnética cardíaca
La localización de la hipertrofia fue predominantemente septal (84,4 % en ecocardiografía y 78,9 % en RMC) y apical (11,2 % y 15 %, respectivamente). No se observaron diferencias significativas en la fracción de eyección del ventrículo izquierdo entre ecocardiografía y RMC (mediana 61 % [RIC 52-70] vs. 66 % [RIC 55-77]; p=0,114).
Entre los 96 pacientes evaluados con RMC, el 81,2 % presentó realce tardío positivo, con predominio de patrón intramiocárdico (92 %). La localización más frecuente fue el septo interventricular (75 %), con distribución parcheada en el 55,4 % y una mediana de fibrosis del 5,1 % (RIC 2,8-17).
Se observó gradiente obstructivo del tracto de salida del ventrículo izquierdo (>30 mmHg) en 55 pacientes (34,3 %). La mediana del gradiente en reposo fue de 43 mmHg (RIC 28-66), aumentando a 62 mmHg (RIC 37-82) con la maniobra de Valsalva. Entre los pacientes sometidos a test genético, el 82,8 % presentó un resultado positivo para variantes patogénicas o probablemente patogénicas (n=53/64). Las variantes más frecuentes se identificaron en los genes sarcoméricos MYH7 (n=21), MYBPC3 (n=13) y TNNT2 (n=6). Se realizó un análisis comparativo entre pacientes sometidos y no sometidos a test genético (Tabla 2). Los pacientes testeados eran significativamente más jóvenes (mediana 37 años [RIC 25-48] vs. 58 años [RIC 45-70]; p < 0,001) y presentaban mayor prevalencia de antecedentes familiares de muerte súbita (25 % vs. 12,5 %; p = 0,043). Asimismo, el score de riesgo de muerte súbita no difirió entre ambos grupos.
Tabla 2
Comparación entre pacientes con y sin test genético
| Variable | Test genético | Sin test genético | p |
|---|---|---|---|
| n=64 | n=96 | ||
| Edad, años (mediana, RIC) | 37 (25-48) | 58 (45-70) | <0, 001 |
| Sexo masculino, n (%) | 38 (59,3) | 58 (60,4) | 0, 401 |
| Antecedente familiar de muerte súbita, n (%) | 16 (25) | 12 (12,5) | 0, 038 |
| Disnea, n (%) | 12 (18,7) | 18 (18,7) | 1, 000 |
| Angina, n (%) | 7 (10,9) | 8 (8,3) | 0, 592 |
| Obstrucción TSVI, n (%) | 15 (23,4) | 21 (21,8) | 0, 818 |
| RMC realizada, n (%) | 33 (51,5) | 63 (65,6) | 0, 564 |
| Fibrosis en RMC, n (%) | 26/31 (83,8) | 52/62 (83,8) | 0, 679 |
| Score ESC riesgo MS, % (mediana, RIC) | 5,8 (3,2-8,1) | 6,5 (3,5-9,0) | 0, 417 |
ESC: European Society of Cardiology; MS: muerte súbita; RIC: rango intercuartílico; RMC: resonancia magnética cardiaca; TSVI: tracto de salida del ventrículo izquierdo
Las principales causas de no realización del estudio fueron la ausencia de cobertura de salud (n=29), la falta de disponibilidad local (n=16) y la decisión del médico tratante (n=15); en un grupo menor se consignaron otros motivos (n=21), mientras que en el resto no se pudo determinar la causa. El estudio familiar se efectuó en el 44,9 % de la cohorte (n=72), empleándose principalmente el ecocardiograma (n=59), seguido del test genético (n=35) y la RMC (n=22).
Respecto al tratamiento, los fármacos más empleados fueron betabloqueantes (42,5 %), inhibidores del sistema renina-angiotensina (16,9 %), bloqueantes cálcicos (15,6 %) y furosemida (11,2 %). La Figura 3 muestra la medicación que los pacientes recibían antes de la primera consulta y la indicada posteriormente según criterio clínico.
Fig. 3
Medicación previa y agregada
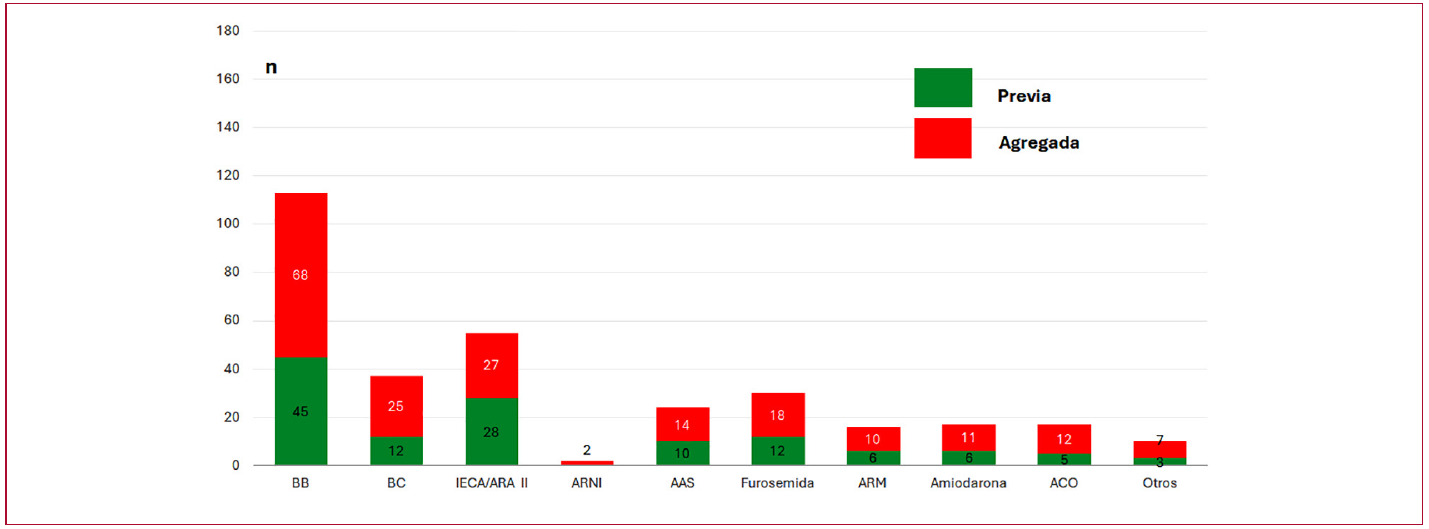
AAS: ácido acetilsalicílico; ACO: anticoagulantes orales; ARA II: antagonistas del receptor de angiotensina II; ARM: antagonistas de los receptores mineralocorticoides; ARNI: inhibidores del receptor de angiotensina/neprilisina BB: betabloqueantes; BC: bloqueantes cálcicos; IECA: inhibidores de la enzima convertidora de angiotensina.
Entre los 20 pacientes con diagnóstico previo de MCH en seguimiento por su cardiólogo tratante, 11 presentaban cardiodesfibrilador implantable (CDI), 4 habían sido sometidos a miectomía, 3 portaban marcapasos y 2 habían recibido ablación septal con alcohol al momento del registro. En el total de la cohorte analizada, 35 pacientes presentaban antecedente de intervenciones terapéuticas posteriores al diagnóstico de MCH, incluyendo 26 implantes de CDI, 4 marcapasos, 4 miectomías septales y 1 dispositivo de resincronización. La miectomía redujo significativamente el gradiente del TSVI en dos tercios de los casos.
De los 26 pacientes que recibieron un CDI durante el seguimiento, se dispuso del puntaje European Society of Cardiology (ESC) en 10 casos y del puntaje American Heart Association (AHA) en 15. La mediana del puntaje ESC fue 6,5 % (RIC 5,1-8,2). Según las categorías de riesgo, el 60 % (6 pacientes) se encontraba en ≥6 % (alto riesgo), 2 pacientes en riesgo intermedio (4-6 %) y 2 en riesgo bajo (<4 %). La mediana del puntaje AHA fue 6,3 (RIC 3,2-9,4). Se documentaron 3 descargas apropiadas (11,5 % de los pacientes con CDI) al momento del registro.
DISCUSIÓN
Este registro constituye el primer estudio nacional que analiza pacientes con MCH atendidos en centros no especializados. Su relevancia radica en que la mayoría de la literatura internacional proviene de centros de referencia, donde el diagnóstico y el manejo se llevan a cabo siguiendo protocolos estandarizados y con amplio acceso a estudios avanzados. (12-16) En contraste, nuestros resultados reflejan la práctica asistencial real en un país con marcada heterogeneidad en la disponibilidad de recursos y el acceso a prácticas especializadas.
Si bien la gran mayoría de cardiólogos que realizaron el seguimiento de estos pacientes se concentra en Buenos Aires y CABA, se incluyeron centros de algunas provincias del interior (Santa Fe, Formosa, Catamarca, Tucumán, Río Negro, Mendoza y Chubut). El diagnóstico de la miocardiopatía hipertrófica continúa siendo un desafío, incluso en entornos con recursos avanzados, debido a su expresión clínica heterogénea y a la superposición con fenocopias. En nuestro registro, el diagnóstico quedó a criterio del cardiólogo tratante y no siempre se apoyó en estudios complementarios de alta complejidad, lo que refleja las condiciones de práctica clínica real en centros no especializados. Esta característica, que podría considerarse una limitación, también otorga valor al estudio, ya que permite identificar las dificultades diagnósticas en la atención cotidiana fuera de los centros de referencia.
La edad media de nuestra cohorte (48 años) fue similar a la reportada en registros internacionales, como el Hypertrophic Cardiomyopathy Registry (HCMR, 49 años),(17) el EURObservational (47 años),(18) el portugués (53 años),(19) el italiano (44 años), (20) y el Sarcomeric Human Cardiomyopathy Registry (SHaRe, 45,8 años).(21) Asimismo, observamos un predominio masculino (60,6 %), comparable al descrito en HCMR (71 %) y SHaRe (63 %). Sin embargo, existe un creciente consenso en que la menor representación femenina refleja un subdiagnóstico más que una verdadera diferencia en la incidencia de la enfermedad.
En relación con los antecedentes familiares de muerte súbita, el 17,5 % de nuestros pacientes presentó este antecedente, proporción similar a la informada en el registro SHaRe. La evaluación sistemática de esta variable constituye un aspecto relevante de nuestro registro, dado su impacto en la estratificación de riesgo en la MCH.
En cuanto al motivo de diagnóstico, el 28,3 % de los casos correspondió a pacientes sintomáticos y el 28,3 % a hallazgos en controles de rutina, proporciones semejantes a las informadas en los registros EURObservational y portugués. En nuestra cohorte, sin embargo, se observó que el 36,4 % de los pacientes con disnea se encontraba en CF III-IV, mientras que en la mayoría de los registros internacionales esta proporción fue considerablemente menor (<10 %), con un claro predominio de pacientes en CF I-II (≈80 %). Esta diferencia podría reflejar un diagnóstico más tardío en nuestro medio, probablemente asociado a limitaciones en el acceso temprano a especialistas y métodos diagnósticos avanzados. (9,10) La localización de la hipertrofia fue predominantemente septal y, en menor medida, apical, en concordancia con lo descrito en la literatura internacional.De igual modo, la prevalencia de fibrosis detectada por RMC fue elevada (81,2 % entre los pacientes evaluados), en línea con lo informado en registros previos. (7) No obstante, este hallazgo debe interpretarse en el contexto de una población seleccionada, ya que la RMC no se realizó de manera sistemática en toda la cohorte, sino en un subgrupo de pacientes con indicación clínica, lo que podría haber sobreestimado su prevalencia. En contraste, la proporción de pacientes sometidos a ecocardiograma de estrés fue marcadamente baja (10,6 %), a pesar de tratarse de una herramienta accesible y fundamental para diferenciar la obstrucción basal de la provocada, ya sea mediante maniobra de Valsalva o durante el ejercicio. Este hallazgo probablemente refleje la ausencia de protocolos estandarizados y la necesidad de una mayor integración de esta herramienta en los circuitos diagnósticos habituales de los centros no especializados. Por su parte, el test genético se realizó en el 40 % de los pacientes, proporción comparable a la del registro portugués (51 %) y algo menor a la del SHaRe (60 %).(19,21) El rendimiento diagnóstico fue elevado (82,8 %), con predominio de variantes sarcoméricas clásicas (MYH7, MYBPC3 y TNNT2). Este hallazgo debe interpretarse en el contexto de un sesgo de selección. En el análisis comparativo, los pacientes sometidos a estudio genético eran significativamente más jóvenes (mediana 37 años [RIC 25-48] vs. 58 años [RIC 45-70]; p < 0,001) y presentaban mayor prevalencia de antecedentes familiares de muerte súbita (25 % vs. 12,5 %; p = 0,043), sin observarse diferencias en otras variables clínicas o morfológicas. Estos hallazgos sugieren que el test fue indicado preferentemente en individuos con mayor sospecha de etiología genética, más que en función de la severidad clínica o del fenotipo estructural. En conjunto, esto explica, al menos en parte, el mayor rendimiento diagnóstico respecto de lo reportado en cohortes no seleccionadas, y sugiere que las diferencias observadas en la utilización de los distintos métodos diagnósticos responden más a variaciones en su integración dentro de la práctica clínica que a su disponibilidad real, lo que identifica oportunidades de mejora en la sistematización del abordaje diagnóstico en los centros no especializados.
Asimismo, la principal limitación en relación con el estudio genético fue la falta de cobertura de salud, lo que restringió no solo el acceso individual, sino también la posibilidad de implementar estrategias de estudio en cascada familiar. En este contexto, solo el 44,9 % de los familiares fue evaluado, predominantemente mediante ecocardiografía, lo que contrasta con lo reportado en registros europeos y norteamericanos.(17-21)
En cuanto al tratamiento, los betabloqueantes fueron la droga más utilizada, en concordancia con lo informado en otros registros. En la cohorte analizada, 26 pacientes presentaban CDI, en el contexto de la evolución de la enfermedad.
La estratificación de riesgo de muerte súbita mostró una mediana del score ESC de 6,5 %, con la mayoría de los pacientes portadores de CDI ubicados en la categoría de alto riesgo (≥6 %). Estos hallazgos concuerdan con las recomendaciones de las guías ESC para la indicación de CDI en prevención primaria.(5)
No obstante, se identificaron implantes en pacientes con riesgo bajo o intermedio, lo que probablemente refleje la consideración de variables clínicas adicionales -como síncope, taquicardia ventricular no sostenida, realce tardío en RMC o antecedentes familiares- en la toma de decisiones. La disponibilidad incompleta de los puntajes de estratificación limitó un análisis más exhaustivo, aunque la tendencia general sugiere que la mayoría de los CDI fueron indicados en pacientes de mayor riesgo.
Estos hallazgos deben interpretarse en el contexto de una cohorte con una evolución clínica prolongada desde el diagnóstico, con una mediana cercana a los 4 años y un amplio rango de seguimiento. Este aspecto podría influir en la frecuencia de intervenciones observadas, incluyendo el implante de CDI, al reflejar distintas etapas evolutivas de la enfermedad más que eventos incidentes en un período de seguimiento definido. La tasa de miectomía fue baja (4 casos al ingreso y 4 durante el seguimiento), lo que probablemente se relaciona con la escasez de centros con experiencia intervencionista en el país.
Nuestros hallazgos ponen en evidencia dos desafíos centrales en el manejo de la MCH en Argentina. En primer lugar, la inequidad en el acceso a estudios complementarios. Aunque la mayoría de los centros dispone de ecocardiografía de estrés, su baja utilización refleja la ausencia de protocolos estandarizados y la necesidad de una mayor integración de esta práctica en los circuitos diagnósticos habituales. En contraste, los estudios genéticos -menos accesibles- se realizaron en una proporción relativamente alta, aunque condicionados por la cobertura del sistema de salud.
En segundo lugar, la difusión e implementación limitada de guías, directrices y algoritmos diagnósticos. La heterogeneidad en la utilización de herramientas básicas sugiere la necesidad de fortalecer la capacitación y la integración de estrategias diagnósticas estandarizadas en los centros no especializados, lo que podría contribuir a reducir el diagnóstico tardío y la mayor proporción de pacientes en CF III-IV observada en nuestro registro.
En este contexto, las diferencias observadas reflejan no solo la heterogeneidad estructural del sistema de salud argentino, sino también inequidades en los circuitos asistenciales, incluyendo la derivación, la oportunidad diagnóstica y la implementación de estrategias de estratificación de riesgo.
A partir de estos hallazgos, se identifican líneas de acción concretas orientadas a mejorar la atención:
-
Desarrollo de guías nacionales actualizadas adaptadas al contexto argentino, que contemplen los recursos disponibles y definan algoritmos prácticos de diagnóstico y tratamiento. En este sentido, no podemos menos que celebrar la aparición del reciente Consenso Argentino de Diagnóstico y Tratamiento de la Miocardiopatía Hipertrófica, con una revisión exhaustiva del tema y recomendaciones acordes al avance científico y la realidad local. (22)
-
Implementación de programas de educación médica continua dirigidos a profesionales de centros no especializados.
-
Creación de redes de referencia y contrarreferencia que faciliten la derivación oportuna de pacientes con indicaciones de intervenciones avanzadas.
-
Promoción de estrategias de equidad en el acceso al test genético y al screening familiar.
En conjunto, nuestros resultados, en concordancia con los grandes registros internacionales en cuanto a características clínicas, ponen de manifiesto diferencias relevantes en la implementación del proceso diagnóstico y terapéutico, aportando una visión del manejo de la MCH en la práctica clínica real y una base para el desarrollo de estrategias y políticas locales orientadas a reducir la brecha entre la evidencia disponible y su aplicación en la práctica cotidiana.
LIMITACIONES
Este estudio presenta algunas limitaciones que deben ser consideradas al interpretar los resultados. En primer lugar, el tamaño muestral y la distribución geográfica de los centros participantes no permiten extrapolar los hallazgos a toda la población nacional. En segundo lugar, la naturaleza retrospectiva y voluntaria del registro introduce potenciales sesgos de selección y de reporte, con posible sobrerrepresentación de pacientes con mayor complejidad clínica o seguimiento más estrecho.
Asimismo, la heterogeneidad en la disponibilidad y utilización de estudios complementarios entre centros y provincias condiciona la comparabilidad interna y limita la estandarización del abordaje diagnóstico. En este contexto, el diagnóstico final de MCH quedó a criterio del cardiólogo tratante, lo que refleja la práctica clínica real, aunque podría haber introducido variabilidad en la definición de casos.
Por otro lado, la ausencia de un seguimiento prospectivo sistemático limitó la evaluación de eventos clínicos en el tiempo, por lo que los hallazgos se interpretan principalmente en términos de antecedentes al momento del registro. Finalmente, la disponibilidad incompleta de estudios clave -como ecocardiografía de estrés, resonancia magnética cardíaca, test genético y puntajes de estratificación de riesgo- restringió el análisis exhaustivo de algunos subgrupos y puede haber condicionado la interpretación de ciertos resultados.
CONCLUSIONES
Este primer registro argentino de pacientes con MCH atendidos en centros no especializados permitió caracterizar de manera integral la población y las estrategias diagnósticas y terapéuticas empleadas en la práctica clínica real. A pesar de las similitudes con los grandes registros internacionales en términos de características clínicas, se identificaron diferencias relevantes en la utilización de estudios complementarios, en la oportunidad diagnóstica y en la implementación de estrategias terapéuticas.
Estos hallazgos ponen de manifiesto inequidades tanto en el acceso a recursos diagnósticos como en los circuitos asistenciales, incluyendo la derivación, la estratificación de riesgo y el abordaje familiar. En este contexto, los resultados del presente estudio aportan evidencia local que permite orientar el desarrollo de estrategias concretas, entre las que se encuentran la capacitación de equipos de salud, la consolidación de redes de referencia y la adaptación de guías internacionales al contexto argentino, con el objetivo de optimizar el diagnóstico y manejo de esta enfermedad a nivel nacional.
Declaración de conflicto de intereses
Los autores declaran que no tienen conflicto de intereses.
(Véanse formularios de conflicto de intereses de los autores en la Web).
Agradecimientos
A los investigadores participantes del registro y a la Sociedad Argentina de Cardiología por su apoyo logístico y académico.
Financiamiento
Los autores declaran que el presente trabajo no contó con ningún tipo de financiamiento público, privado o institucional.